

Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples
- PMID: 32605661
- PMCID: PMC7325194
- DOI: 10.1186/s13073-020-00751-4
Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples
Abstract
Background: COVID-19 (coronavirus disease 2019) has caused a major epidemic worldwide; however, much is yet to be known about the epidemiology and evolution of the virus partly due to the scarcity of full-length SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) genomes reported. One reason is that the challenges underneath sequencing SARS-CoV-2 directly from clinical samples have not been completely tackled, i.e., sequencing samples with low viral load often results in insufficient viral reads for analyses.
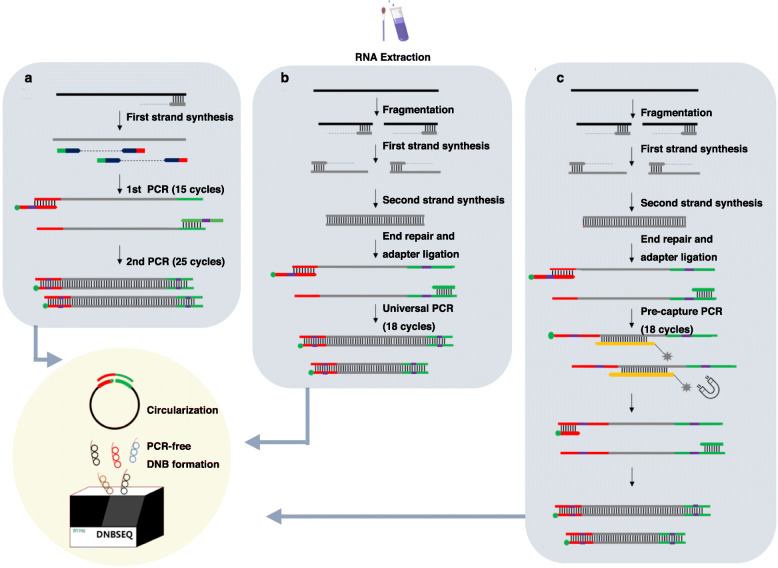
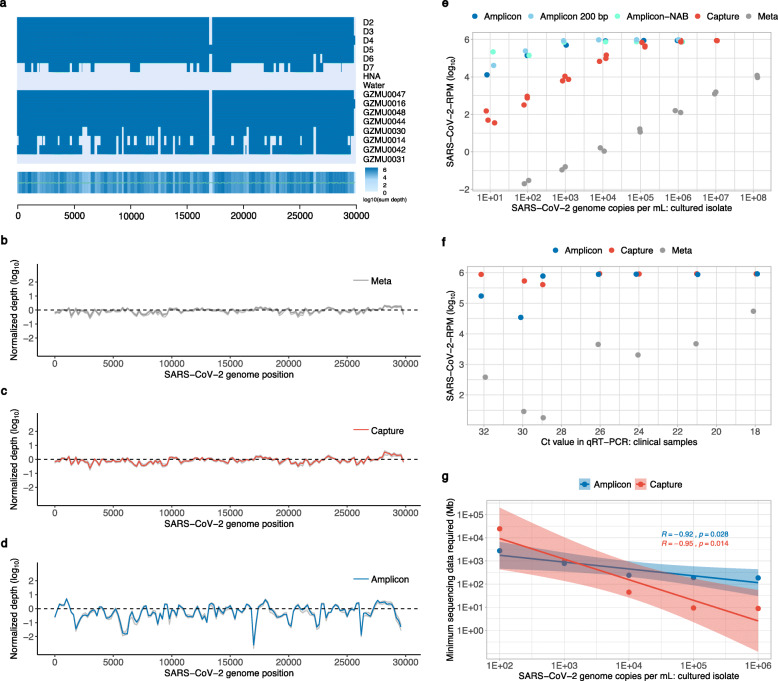
Methods: We applied a novel multiplex PCR amplicon (amplicon)-based and hybrid capture (capture)-based sequencing, as well as ultra-high-throughput metatranscriptomic (meta) sequencing in retrieving complete genomes, inter-individual and intra-individual variations of SARS-CoV-2 from serials dilutions of a cultured isolate, and eight clinical samples covering a range of sample types and viral loads. We also examined and compared the sensitivity, accuracy, and other characteristics of these approaches in a comprehensive manner.
Results: We demonstrated that both amplicon and capture methods efficiently enriched SARS-CoV-2 content from clinical samples, while the enrichment efficiency of amplicon outran that of capture in more challenging samples. We found that capture was not as accurate as meta and amplicon in identifying between-sample variations, whereas amplicon method was not as accurate as the other two in investigating within-sample variations, suggesting amplicon sequencing was not suitable for studying virus-host interactions and viral transmission that heavily rely on intra-host dynamics. We illustrated that meta uncovered rich genetic information in the clinical samples besides SARS-CoV-2, providing references for clinical diagnostics and therapeutics. Taken all factors above and cost-effectiveness into consideration, we proposed guidance for how to choose sequencing strategy for SARS-CoV-2 under different situations.
Conclusions: This is, to the best of our knowledge, the first work systematically investigating inter- and intra-individual variations of SARS-CoV-2 using amplicon- and capture-based whole-genome sequencing, as well as the first comparative study among multiple approaches. Our work offers practical solutions for genome sequencing and analyses of SARS-CoV-2 and other emerging viruses.
Keywords: COVID-19; Emerging infectious diseases; Genomic surveillance; Hybrid capture; Metatranscriptomic sequencing; Multiplex PCR; Quasispecies; Virus evolution; iSNV.
Conflict of interest statement
L.Y., Y.Z., F.C., and X.X. have applied for a patent relating to the amplicon-based method, and the details can be found below:
PCR primer pair and application thereof
Patent applicant: MGI Tech Co., Ltd
Name of inventor(s): Lin Yang, Ya Gao, Guodong Huang, Yicong Wang, Yuqian Wang, Yanyan Zhang, Fang Chen, Na Zhong, Hui Jiang, Xun Xu
Application number: PCT/CN2017/089195
The remaining authors declare that they have no competing interests.
Figures


References
-
- WHO: Coronavirus disease (COVID-2019) situation report - 54. World Health Organization; 2020.
Publication types
MeSH terms
Substances
Grants and funding
- 2019YFC1200501/State Key Research Development Program of China/International
- 2018ZX10301101-004/National Major Project for Control and Prevention of Infectious Disease in China/International
- 2020YFC0841400/the emergency grants for prevention and control of SARS-CoV-2 of Ministry of Science and Technology/International
- 2020B111107001, 2020B111108001, 2018B020207013/the emergency grants for prevention and control of SARS-CoV-2 of Guangdong Province/International
- 2017B030301011/Guangdong Provincial Key Laboratory of Genome Read and Write/International
- 2017B090904014/Guangdong Provincial Academician Workstation of BGI Synthetic Genomics Clinical Animal Diseases (CN)/International
- DRC-SZ[2016]884/Shenzhen Engineering Laboratory for Innovative Molecular Diagnostics/International
- 2017ZX10303406/National Science and Technology Major Project of China/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
