

Diagnosis and Screening of Patients with Fabry Disease
- PMID: 32606714
- PMCID: PMC7319521
- DOI: 10.2147/TCRM.S247814
Diagnosis and Screening of Patients with Fabry Disease
Abstract
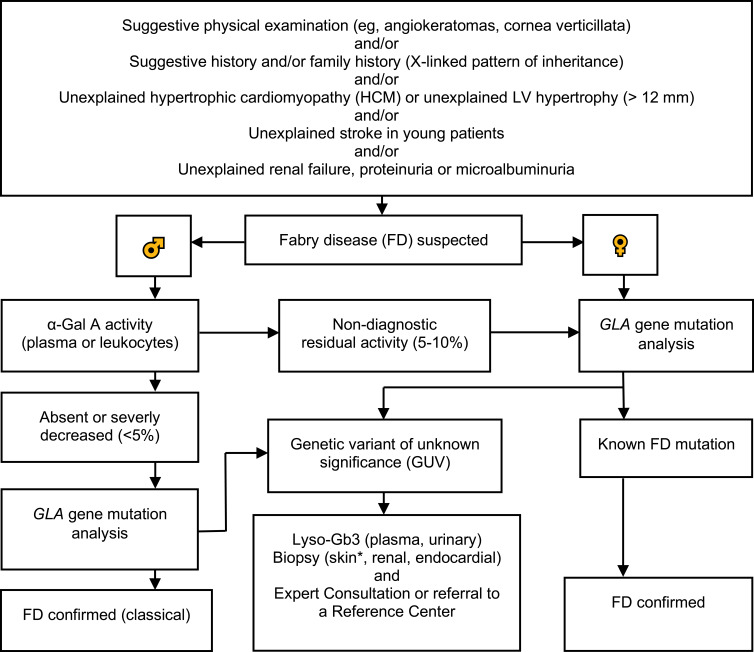
Fabry disease (FD) is an X-linked lysosomal storage disorder caused by absence or deficient activity of α-galactosidase A (α-Gal A) due to mutations in the α-galactosidase A gene (GLA), leading to progressive accumulation of globotriaosylceramide (Gb3) in tissues and organs including heart, kidney, the eyes, vascular endothelium, the nervous system and the skin. Cardiac involvement is leading to fatal complications and reduced life expectancy. FD is treatable with disease-specific treatment (enzyme replacement therapy (ERT) or with chaperone therapy). Therefore, the early diagnosis of FD is crucial for reducing the morbidity and mortality. Screening of high-risk populations (eg, patients with unexplained left ventricular hypertrophy (LVH), young patients with unexplained stroke, and patients with unexplained renal failure proteinuria or microalbuminuria) yields good results. The diagnostic algorithm is gender-specific. Initially, the measurement of α-Gal A activity is recommended in males, and optionally in females. In males with non-diagnostic residual activity (5-10%) activity, genetic testing is afterwards done for confirming the diagnosis. In fact, diagnosis of FD is not possible without genetic testing for both males and females. Globotriaosysphingosine (lyso-Gb3) for identification of atypical FD variants and high- sensitive troponin T (hsTNT) for identification of cardiac involvement are also important diagnostic biomarkers. The aim of this review was to provide an update on diagnosis and screening of patients with FD.
Keywords: algorithm; genetics; heart failure; hypertrophic cardiomyopathy; metabolic disease; proteinuria.
© 2020 Vardarli et al.
Conflict of interest statement
Dr. Vardarli and Dr. Rischpler report no conflicts of interest in this work. Dr. Herrmann reports personal fees from Bayer, stock options (less than 1%) from Sofie Biosciences, personal fees from SIRTEX, non-financial support from ABX, personal fees from Adacap, personal fees from Curium, personal fees from Endocyte, grants and personal fees from BTG, personal fees from IPSEN, personal fees from Siemens Healthineers, personal fees from GE Healthcare, personal fees from Amgen, personal fees from Novartis, personal fees from Y-mAbs, outside the submitted work. Dr. Weidemann has received research grants from Genzyme and Shire and speaker honoraria from Amicus, Genzyme, and Shire.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
