

Reduced replication origin licensing selectively kills KRAS-mutant colorectal cancer cells via mitotic catastrophe
- PMID: 32612138
- PMCID: PMC7330027
- DOI: 10.1038/s41419-020-2704-9
Reduced replication origin licensing selectively kills KRAS-mutant colorectal cancer cells via mitotic catastrophe
Abstract
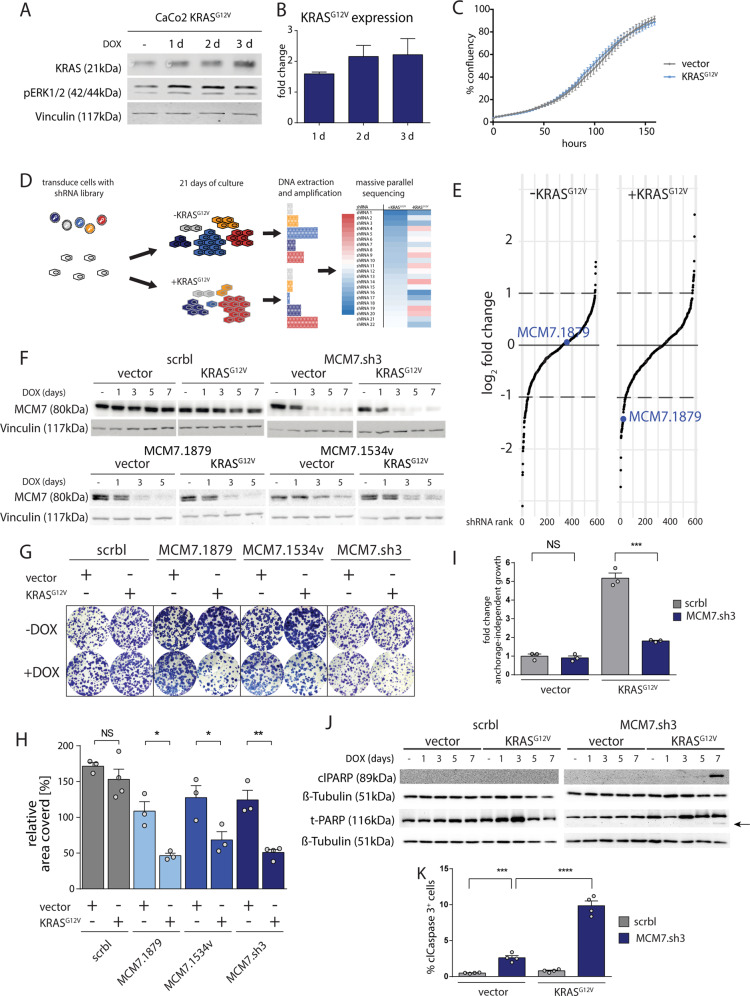
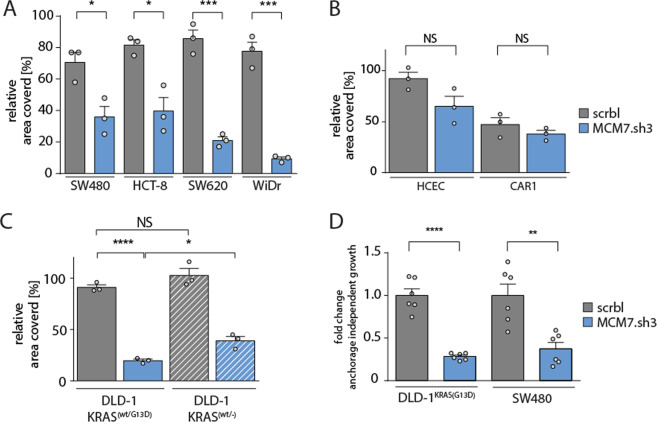
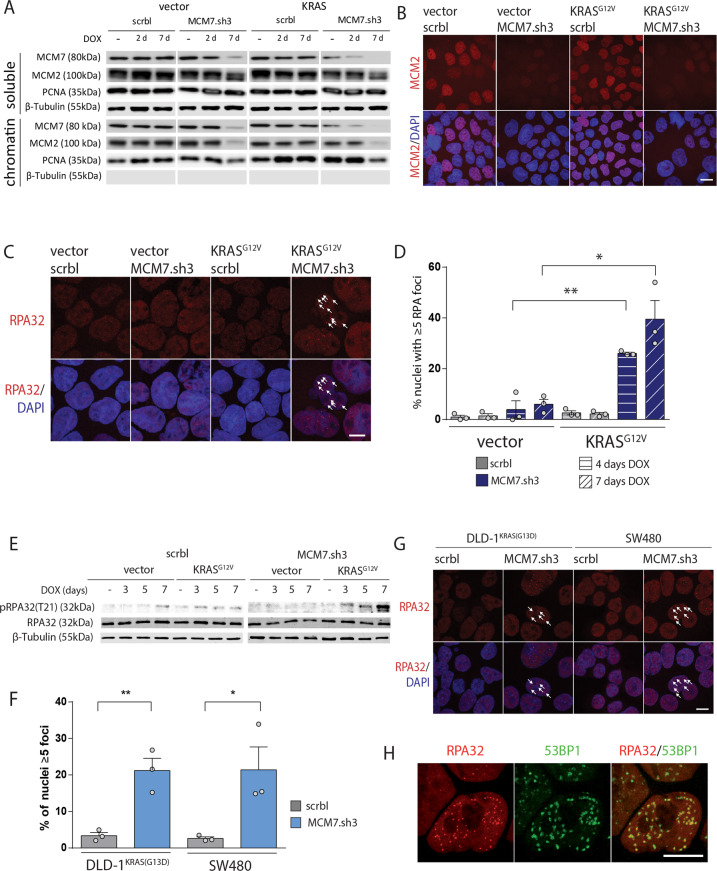
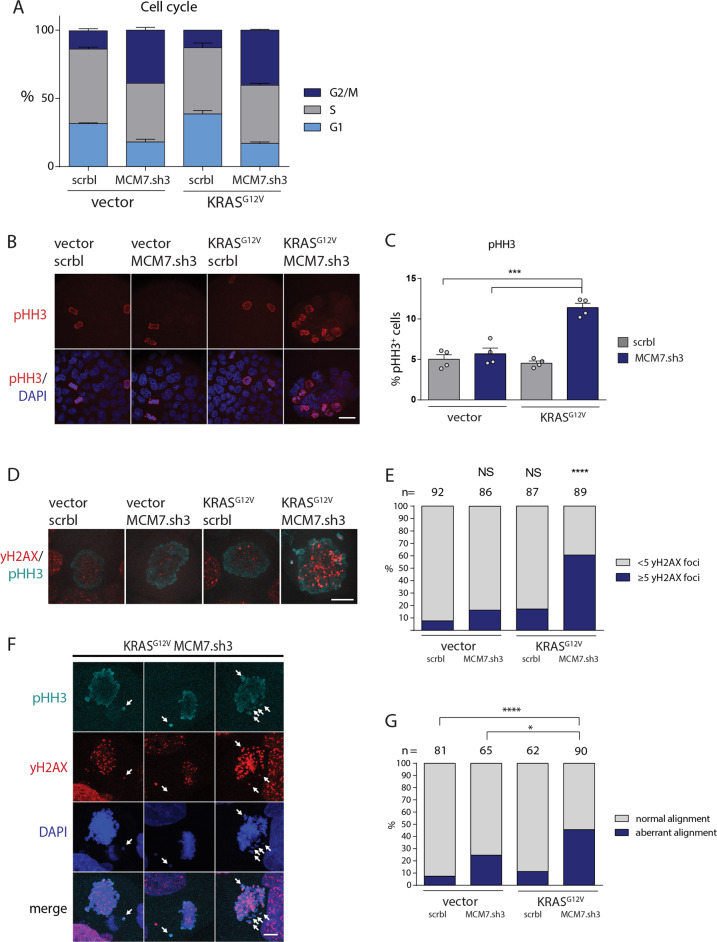
To unravel vulnerabilities of KRAS-mutant CRC cells, a shRNA-based screen specifically inhibiting MAPK pathway components and targets was performed in CaCo2 cells harboring conditional oncogenic KRASG12V. The custom-designed shRNA library comprised 121 selected genes, which were previously identified to be strongly regulated in response to MEK inhibition. The screen showed that CaCo2 cells expressing KRASG12V were sensitive to the suppression of the DNA replication licensing factor minichromosome maintenance complex component 7 (MCM7), whereas KRASwt CaCo2 cells were largely resistant to MCM7 suppression. Similar results were obtained in an isogenic DLD-1 cell culture model. Knockdown of MCM7 in a KRAS-mutant background led to replication stress as indicated by increased nuclear RPA focalization. Further investigation showed a significant increase in mitotic cells after simultaneous MCM7 knockdown and KRASG12V expression. The increased percentage of mitotic cells coincided with strongly increased DNA damage in mitosis. Taken together, the accumulation of DNA damage in mitotic cells is due to replication stress that remained unresolved, which results in mitotic catastrophe and cell death. In summary, the data show a vulnerability of KRAS-mutant cells towards suppression of MCM7 and suggest that inhibiting DNA replication licensing might be a viable strategy to target KRAS-mutant cancers.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Enhanced dependency of KRAS-mutant colorectal cancer cells on RAD51-dependent homologous recombination repair identified from genetic interactions in Saccharomyces cerevisiae.Mol Oncol. 2017 May;11(5):470-490. doi: 10.1002/1878-0261.12040. Epub 2017 Mar 27. Mol Oncol. 2017. PMID: 28173629 Free PMC article.
-
DNA replication stress and mitotic catastrophe mediate sotorasib addiction in KRASG12C-mutant cancer.J Biomed Sci. 2023 Jun 29;30(1):50. doi: 10.1186/s12929-023-00940-4. J Biomed Sci. 2023. PMID: 37386628 Free PMC article.
-
Clinical Role of ASCT2 (SLC1A5) in KRAS-Mutated Colorectal Cancer.Int J Mol Sci. 2017 Jul 27;18(8):1632. doi: 10.3390/ijms18081632. Int J Mol Sci. 2017. PMID: 28749408 Free PMC article.
-
Pyruvate dehydrogenase kinase 4 exhibits a novel role in the activation of mutant KRAS, regulating cell growth in lung and colorectal tumour cells.Oncogene. 2017 Nov 2;36(44):6164-6176. doi: 10.1038/onc.2017.224. Epub 2017 Jul 10. Oncogene. 2017. PMID: 28692044 Free PMC article.
-
Review: KRAS mutations are influential in driving hepatic metastases and predicting outcome in colorectal cancer.Chin Clin Oncol. 2019 Oct;8(5):53. doi: 10.21037/cco.2019.08.16. Chin Clin Oncol. 2019. PMID: 31597434 Review.
Cited by
-
Targeting the DNA replication stress phenotype of KRAS mutant cancer cells.Sci Rep. 2021 Feb 11;11(1):3656. doi: 10.1038/s41598-021-83142-y. Sci Rep. 2021. PMID: 33574444 Free PMC article.
-
Identification of ATP-Competitive Human CMG Helicase Inhibitors for Cancer Intervention that Disrupt CMG-Replisome Function.Mol Cancer Ther. 2024 Nov 4;23(11):1568-1585. doi: 10.1158/1535-7163.MCT-23-0904. Mol Cancer Ther. 2024. PMID: 38982858 Free PMC article.
-
Small Molecule Inhibitor Targeting CDT1/Geminin Protein Complex Promotes DNA Damage and Cell Death in Cancer Cells.Front Pharmacol. 2022 Apr 25;13:860682. doi: 10.3389/fphar.2022.860682. eCollection 2022. Front Pharmacol. 2022. PMID: 35548337 Free PMC article.
-
BCL6 is regulated by the MAPK/ELK1 axis and promotes KRAS-driven lung cancer.J Clin Invest. 2022 Nov 15;132(22):e161308. doi: 10.1172/JCI161308. J Clin Invest. 2022. PMID: 36377663 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
