

Terabase-scale metagenome coassembly with MetaHipMer
- PMID: 32612216
- PMCID: PMC7329831
- DOI: 10.1038/s41598-020-67416-5
Terabase-scale metagenome coassembly with MetaHipMer
Abstract
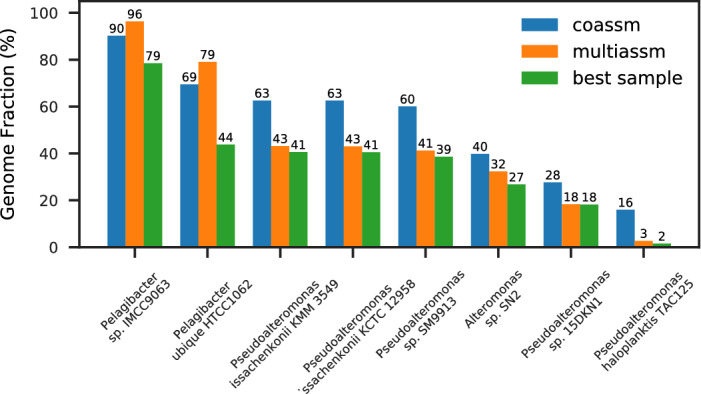
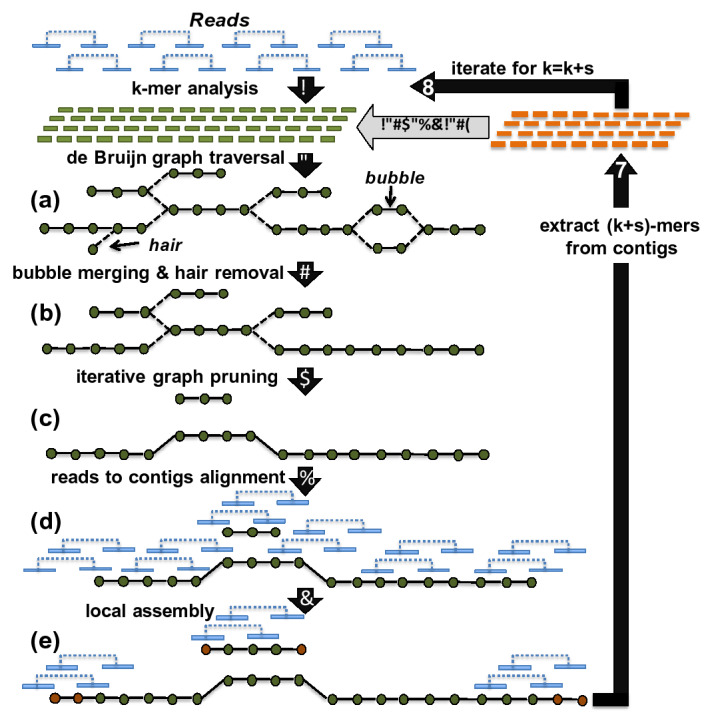
Metagenome sequence datasets can contain terabytes of reads, too many to be coassembled together on a single shared-memory computer; consequently, they have only been assembled sample by sample (multiassembly) and combining the results is challenging. We can now perform coassembly of the largest datasets using MetaHipMer, a metagenome assembler designed to run on supercomputers and large clusters of compute nodes. We have reported on the implementation of MetaHipMer previously; in this paper we focus on analyzing the impact of very large coassembly. In particular, we show that coassembly recovers a larger genome fraction than multiassembly and enables the discovery of more complete genomes, with lower error rates, whereas multiassembly recovers more dominant strain variation. Being able to coassemble a large dataset does not preclude one from multiassembly; rather, having a fast, scalable metagenome assembler enables a user to more easily perform coassembly and multiassembly, and assemble both abundant, high strain variation genomes, and low-abundance, rare genomes. We present several assemblies of terabyte datasets that could never be coassembled before, demonstrating MetaHipMer's scaling power. MetaHipMer is available for public use under an open source license and all datasets used in the paper are available for public download.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Royalty, T.M. & Steen, A.D. Simulation-based approaches to characterize the effect of sequencing depth on the quantity and quality of metagenome-assembled genomes. bioRxiv 356840 (2018).
