

Structural cells are key regulators of organ-specific immune responses
- PMID: 32612232
- PMCID: PMC7610345
- DOI: 10.1038/s41586-020-2424-4
Structural cells are key regulators of organ-specific immune responses
Abstract
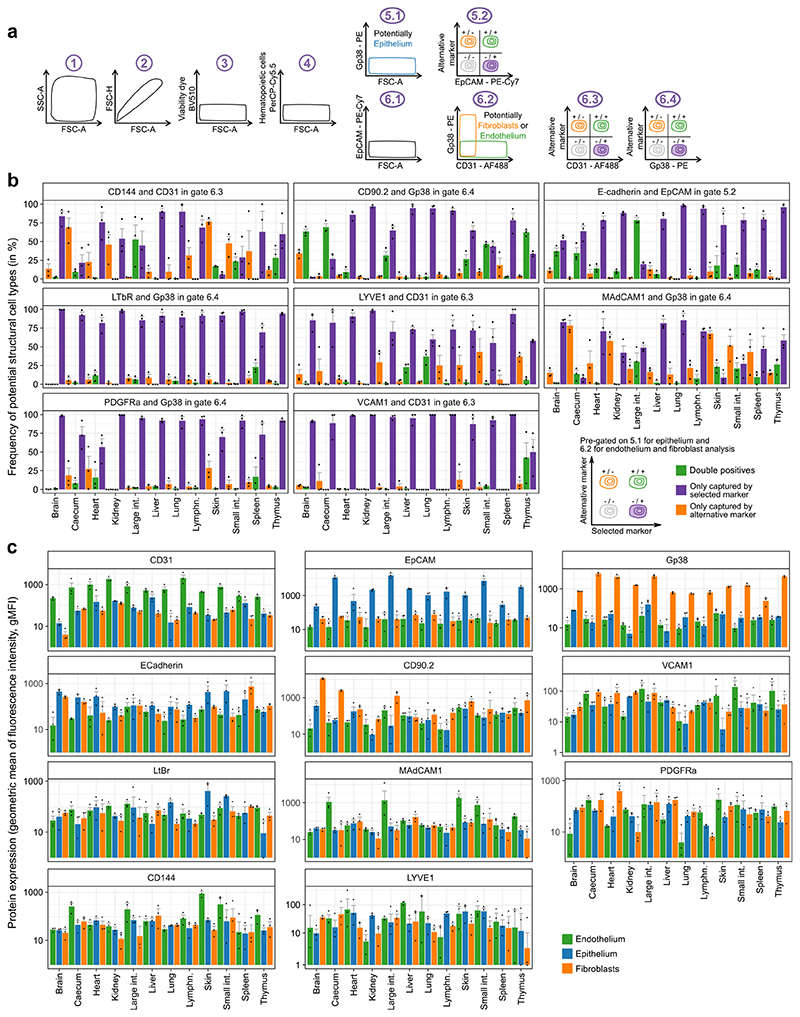
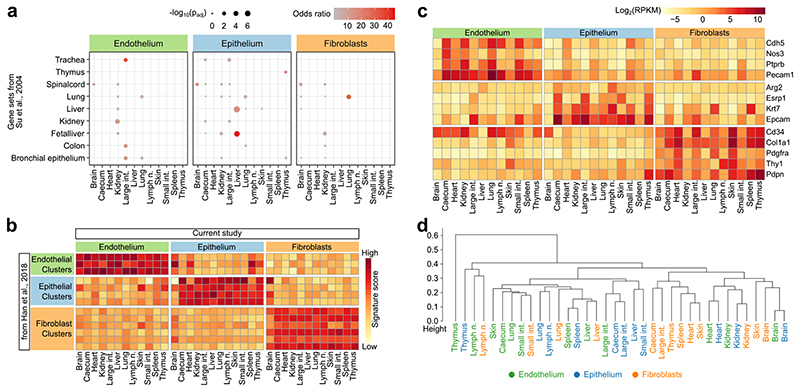
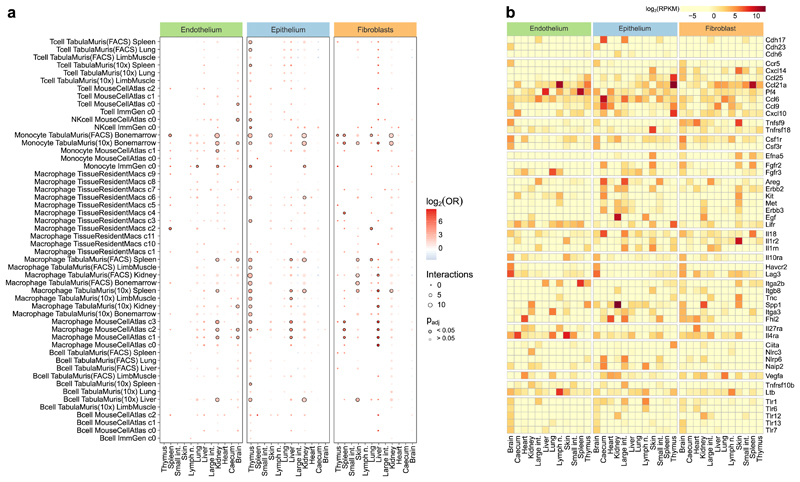
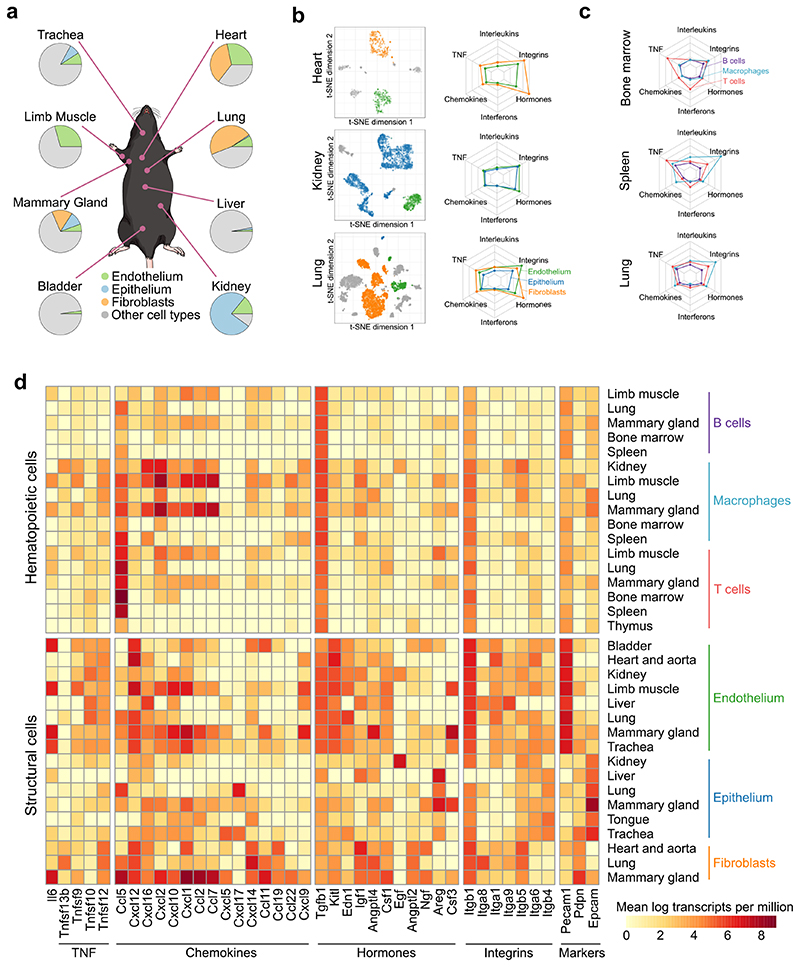
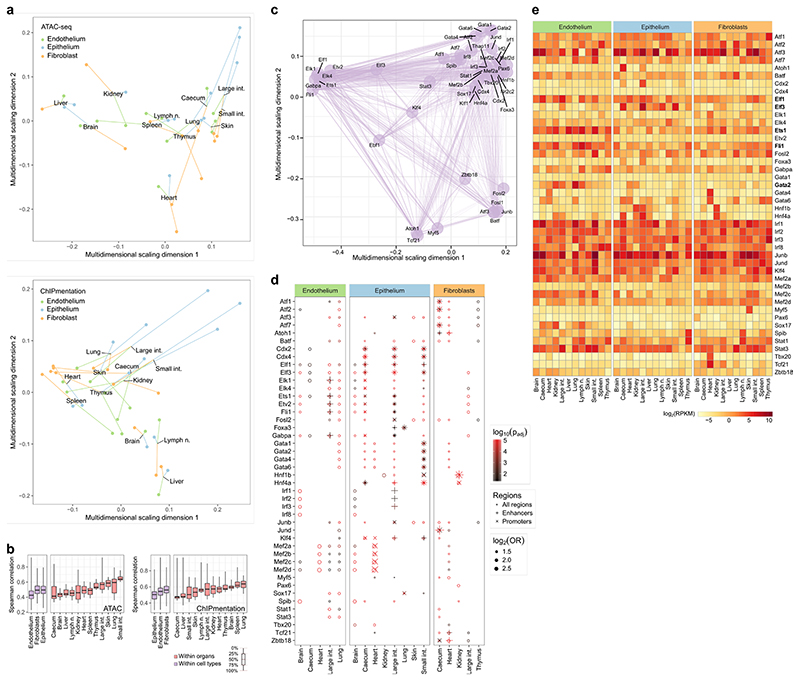
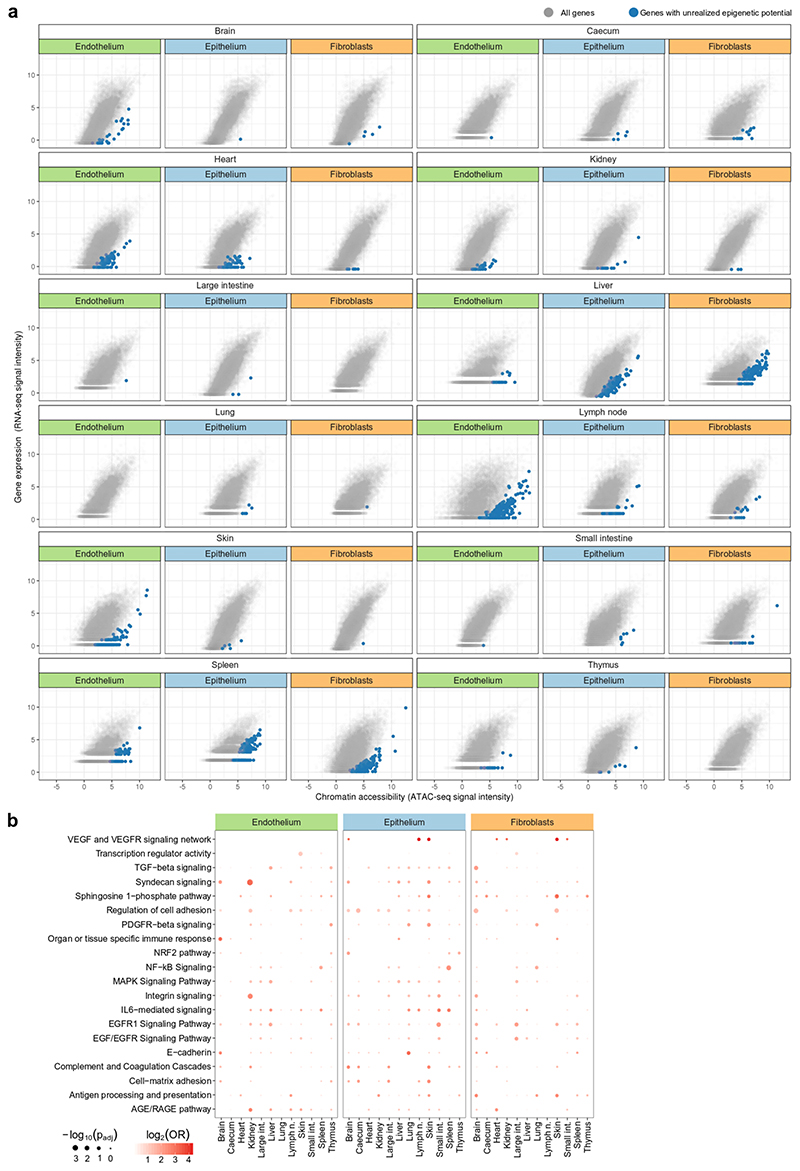
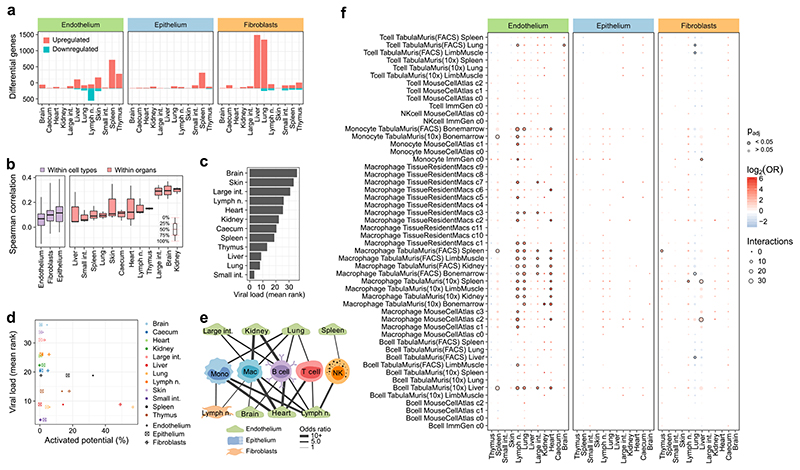
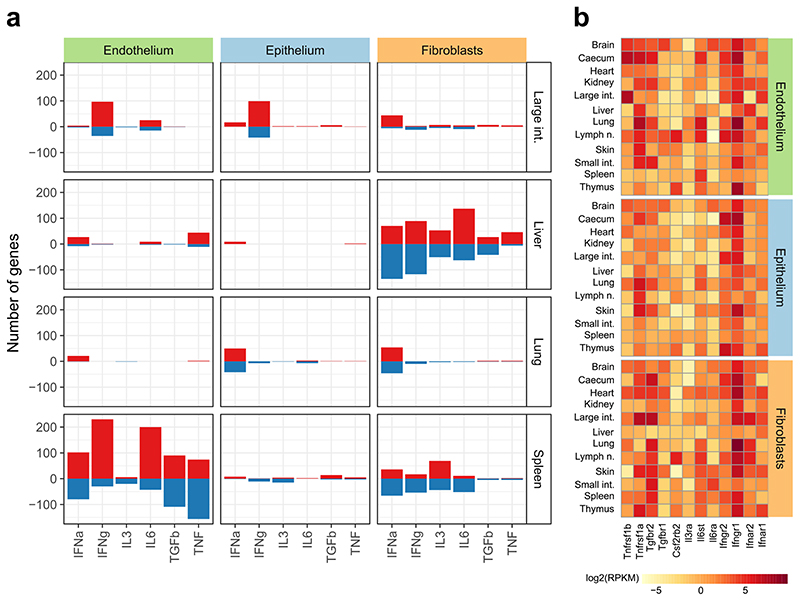
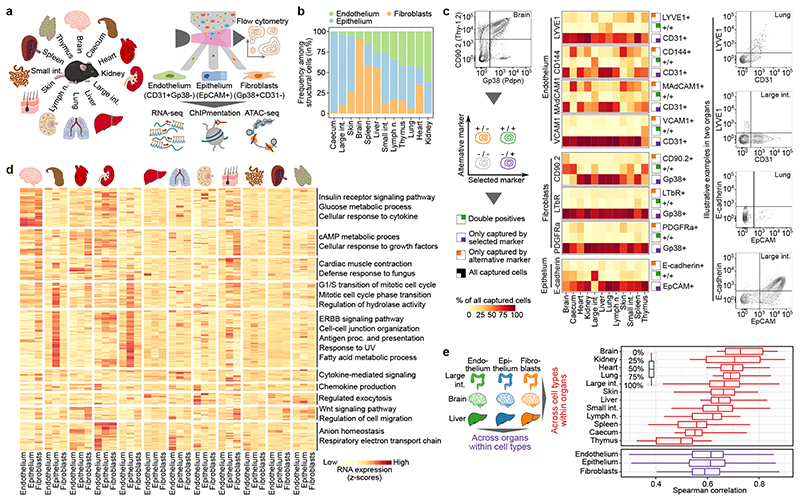
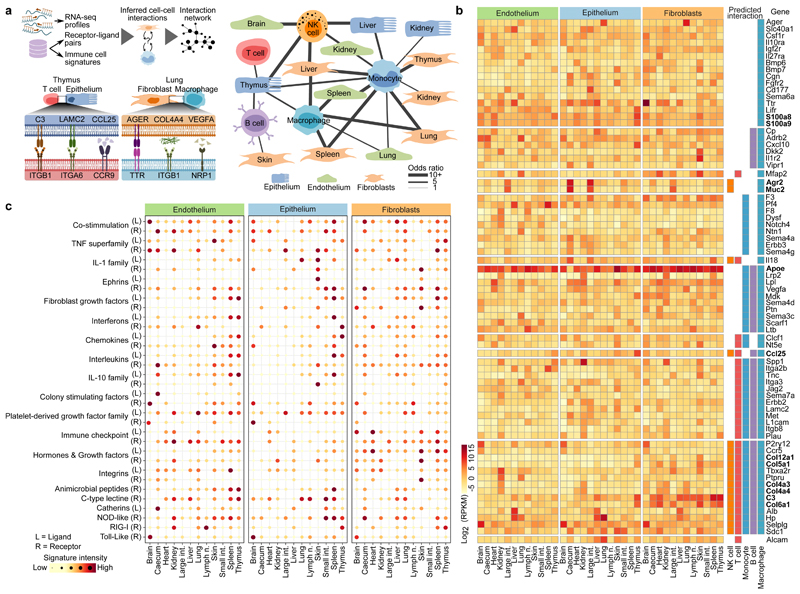
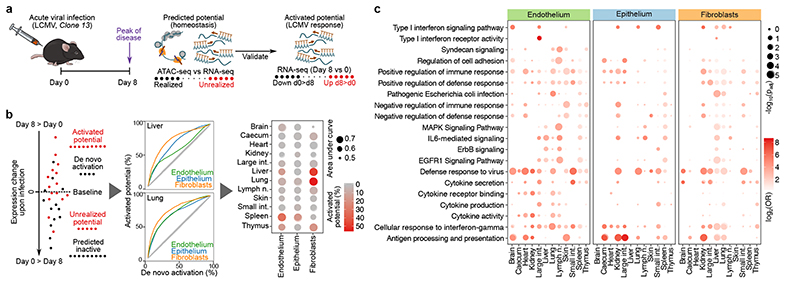
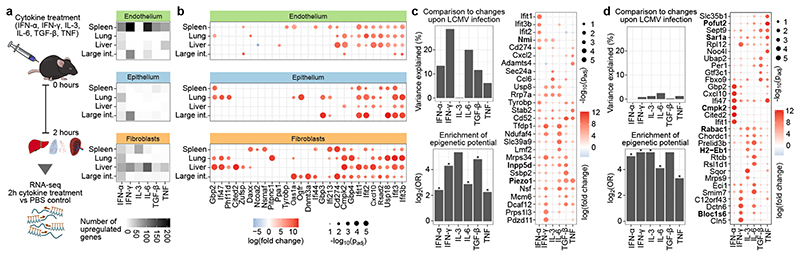
The mammalian immune system implements a remarkably effective set of mechanisms for fighting pathogens1. Its main components are haematopoietic immune cells, including myeloid cells that control innate immunity, and lymphoid cells that constitute adaptive immunity2. However, immune functions are not unique to haematopoietic cells, and many other cell types display basic mechanisms of pathogen defence3-5. To advance our understanding of immunology outside the haematopoietic system, here we systematically investigate the regulation of immune genes in the three major types of structural cells: epithelium, endothelium and fibroblasts. We characterize these cell types across twelve organs in mice, using cellular phenotyping, transcriptome sequencing, chromatin accessibility profiling and epigenome mapping. This comprehensive dataset revealed complex immune gene activity and regulation in structural cells. The observed patterns were highly organ-specific and seem to modulate the extensive interactions between structural cells and haematopoietic immune cells. Moreover, we identified an epigenetically encoded immune potential in structural cells under tissue homeostasis, which was triggered in response to systemic viral infection. This study highlights the prevalence and organ-specific complexity of immune gene activity in non-haematopoietic structural cells, and it provides a high-resolution, multi-omics atlas of the epigenetic and transcriptional networks that regulate structural cells in the mouse.
Conflict of interest statement
Supplementary Information is available for this paper.
Figures




Comment in
-
An antiviral response beyond immune cells.Nature. 2020 Jul;583(7815):206-207. doi: 10.1038/d41586-020-01916-2. Nature. 2020. PMID: 32623443 No abstract available.
-
A gene atlas of 'structural immunity'.Nat Rev Immunol. 2020 Sep;20(9):518-519. doi: 10.1038/s41577-020-0398-y. Nat Rev Immunol. 2020. PMID: 32661408 No abstract available.
-
Organ immune responses - don't forget the structural cells.Nat Rev Nephrol. 2020 Oct;16(10):570-571. doi: 10.1038/s41581-020-00334-x. Nat Rev Nephrol. 2020. PMID: 32792669 No abstract available.
-
Mucosal epithelial cells: the initial sentinels and responders controlling and regulating immune responses to viral infections.Cell Mol Immunol. 2021 Jul;18(7):1628-1630. doi: 10.1038/s41423-021-00650-7. Epub 2021 Apr 29. Cell Mol Immunol. 2021. PMID: 33927362 Free PMC article. No abstract available.
-
Structural Cells as Key Regulators of Organ-specific Immunity.Transplantation. 2021 Jun 1;105(6):1137-1139. doi: 10.1097/TP.0000000000003572. Transplantation. 2021. PMID: 34048416 No abstract available.
References
-
- Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. 2019
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
