

Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling
- PMID: 32613204
- PMCID: PMC7328899
- DOI: 10.1038/s43018-019-0018-6
Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling
Abstract
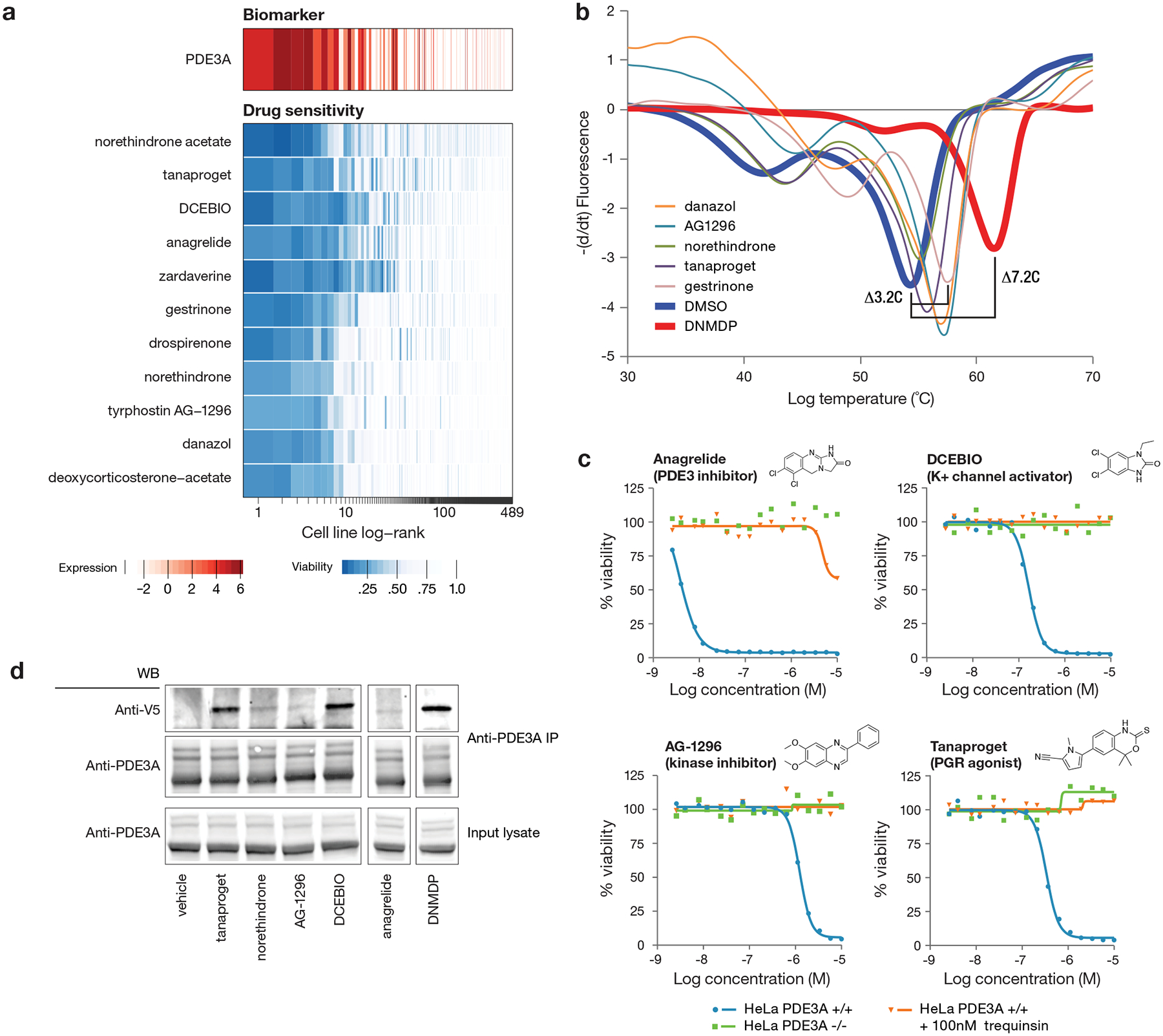
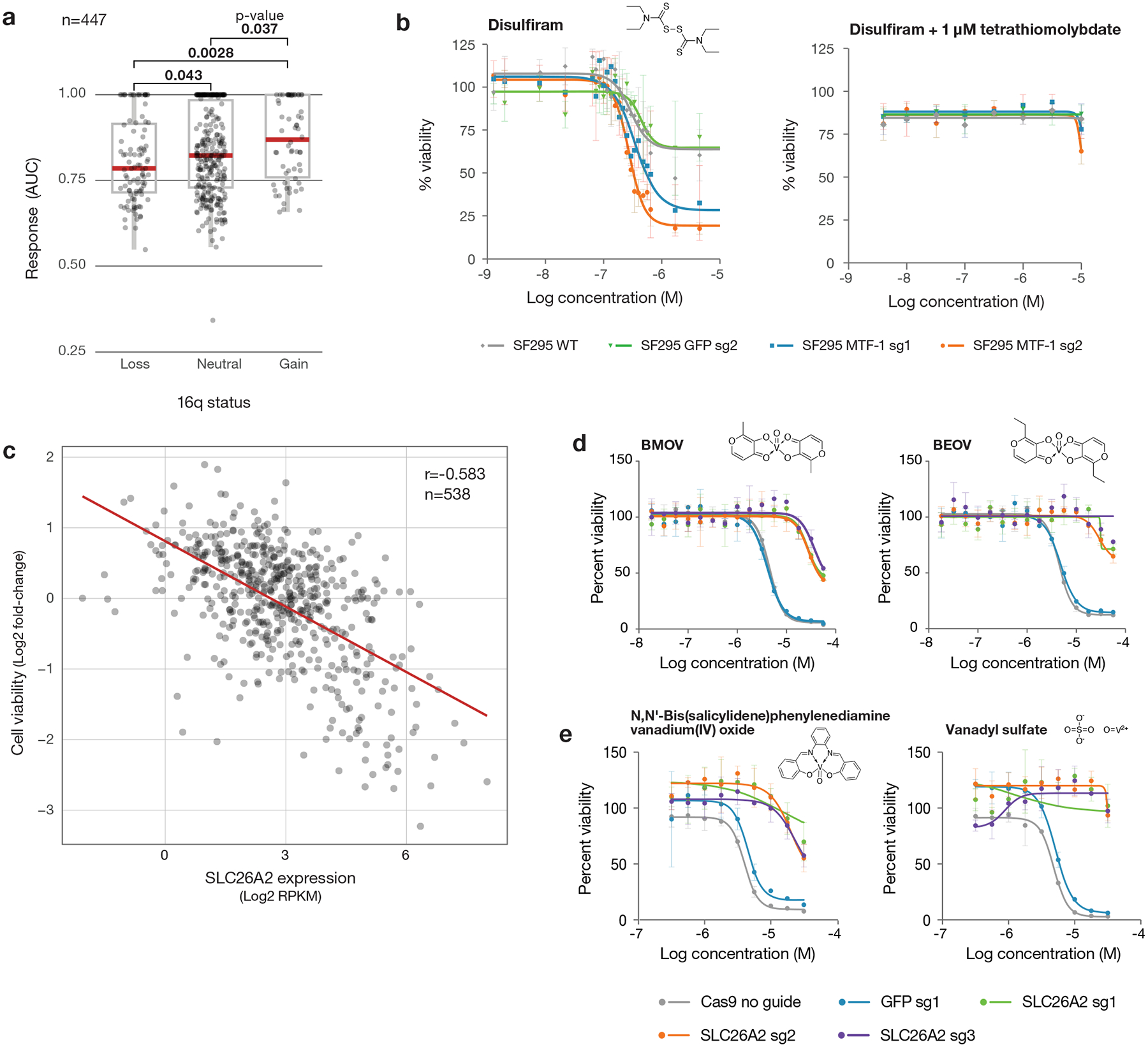
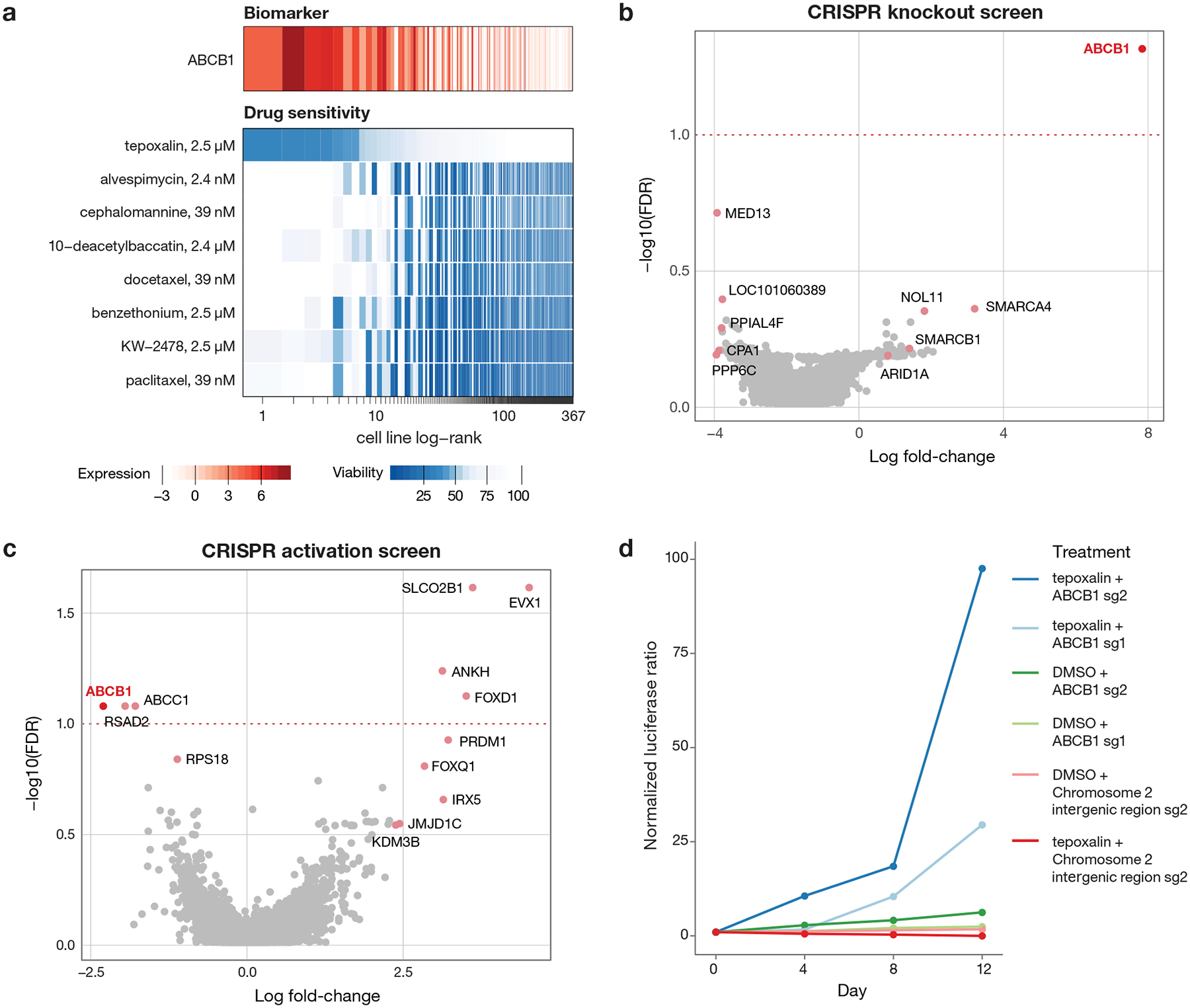
Anti-cancer uses of non-oncology drugs have occasionally been found, but such discoveries have been serendipitous. We sought to create a public resource containing the growth inhibitory activity of 4,518 drugs tested across 578 human cancer cell lines. We used PRISM, a molecular barcoding method, to screen drugs against cell lines in pools. An unexpectedly large number of non-oncology drugs selectively inhibited subsets of cancer cell lines in a manner predictable from the cell lines' molecular features. Our findings include compounds that killed by inducing PDE3A-SLFN12 complex formation; vanadium-containing compounds whose killing depended on the sulfate transporter SLC26A2; the alcohol dependence drug disulfiram, which killed cells with low expression of metallothioneins; and the anti-inflammatory drug tepoxalin, which killed via the multi-drug resistance protein ABCB1. The PRISM drug repurposing resource (https://depmap.org/repurposing) is a starting point to develop new oncology therapeutics, and more rarely, for potential direct clinical translation.
Conflict of interest statement
COMPETING INTERESTS S.M.C, X.W., H.G, M.M., A.S., and T.R.G receive research funding unrelated to this project from Bayer HealthCare. M.M receives research funding from Ono and serves as a scientific advisory board and consultant for OrigiMed. M.M. has patents licensed to LabCorp and Bayer. M.M. and T.R.G. were formerly consultants and equity holders in Foundation Medicine, acquired by Roche. J.A.B. is an employee and shareholder of Vertex Pharmaceuticals. J.G.D. and A.T. consult for Tango Therapeutics. T.R.G. is a consultant to GlaxoSmithKline and is a founder of Sherlock Biosciences. Patent applications for the drug uses detailed in this manuscript have been filed. Other authors declare no competing interests.
Figures
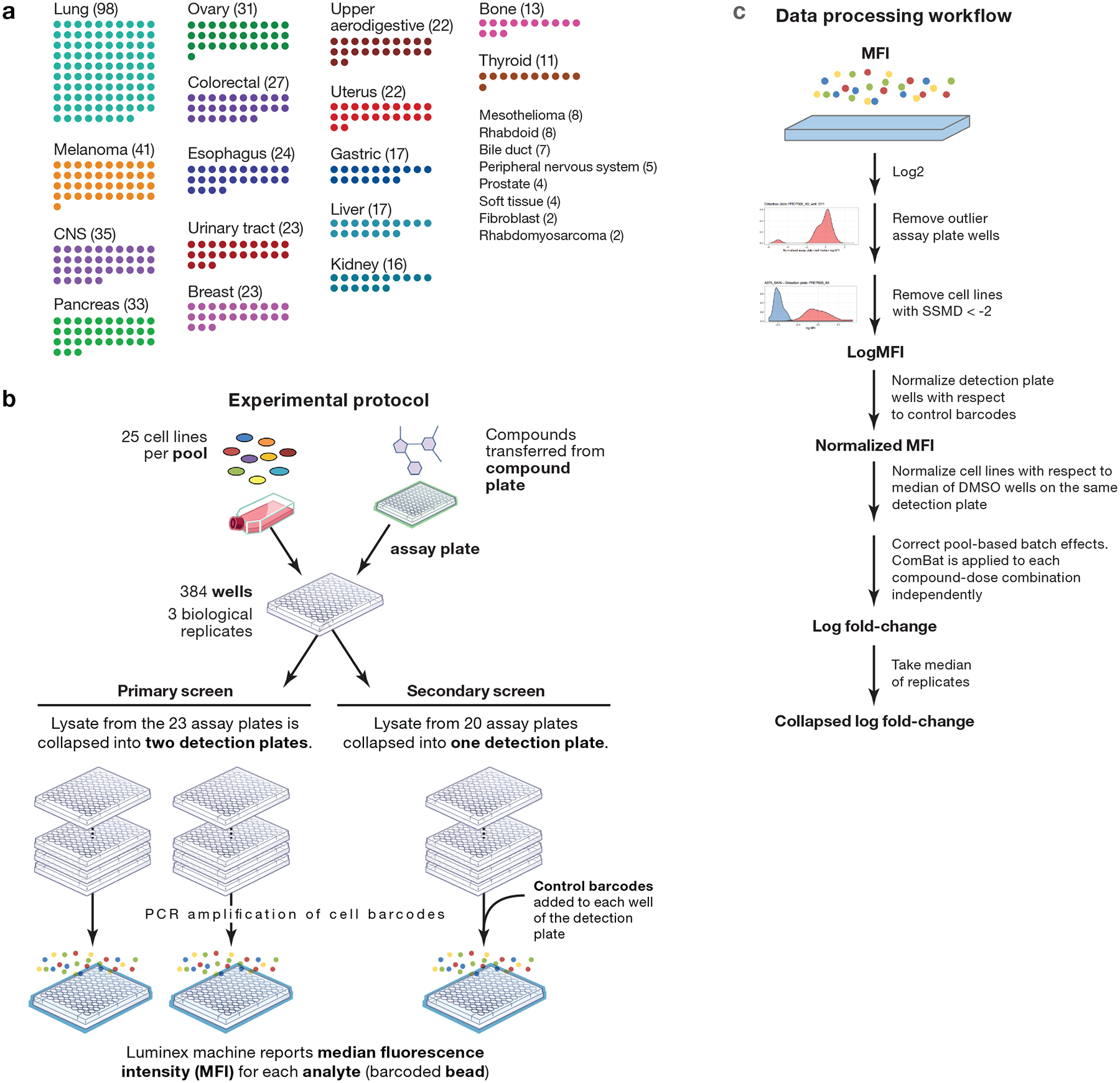
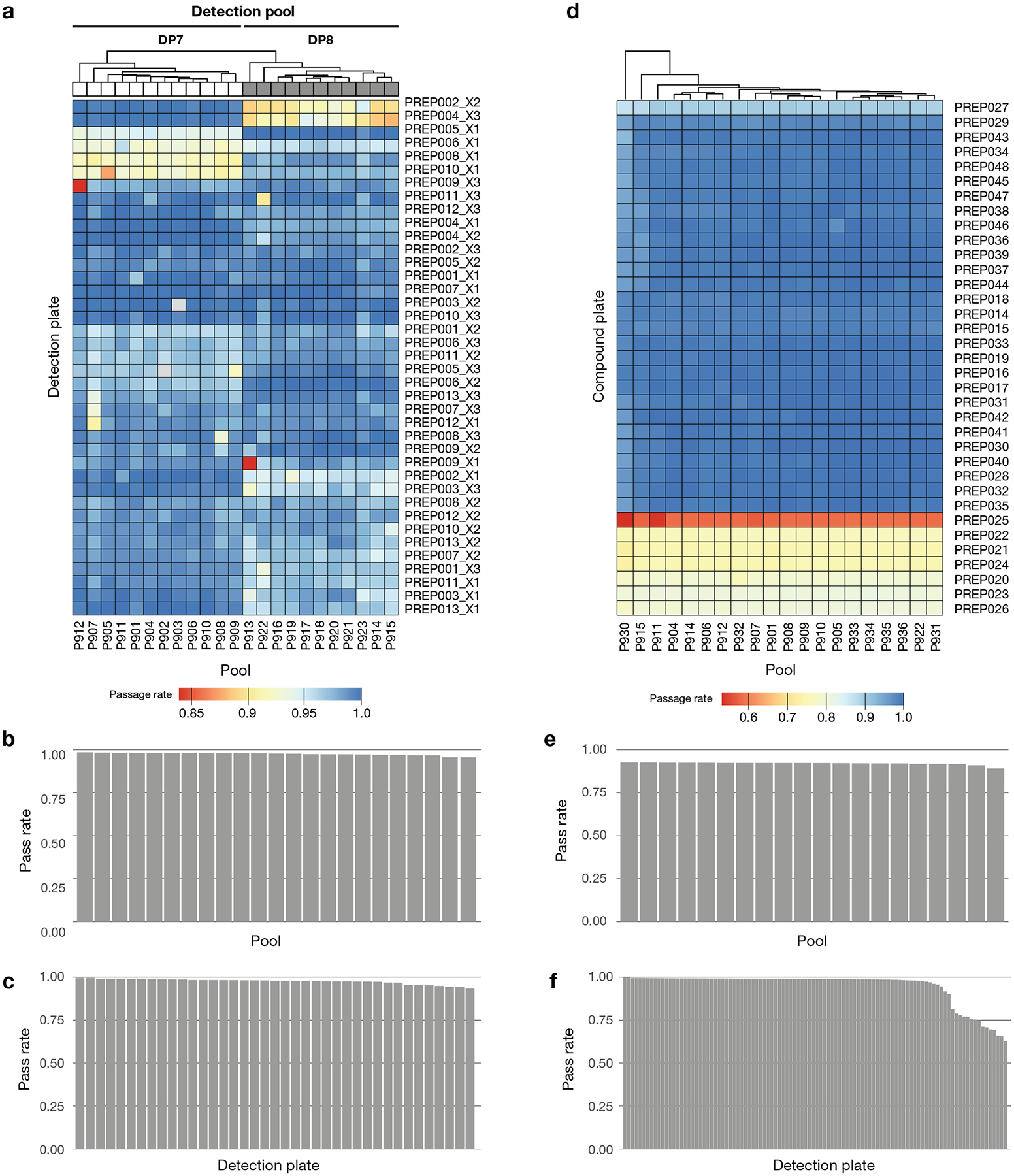
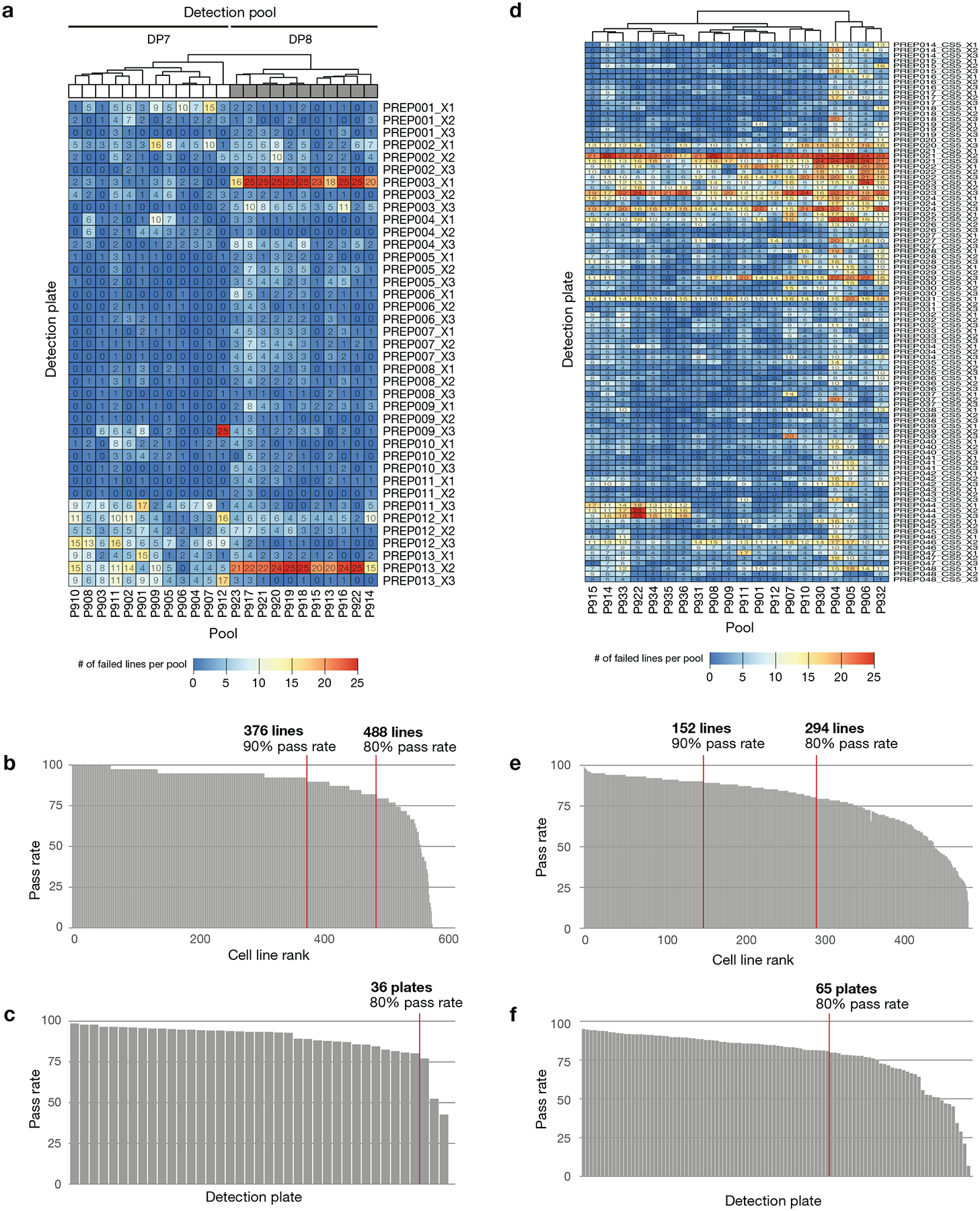
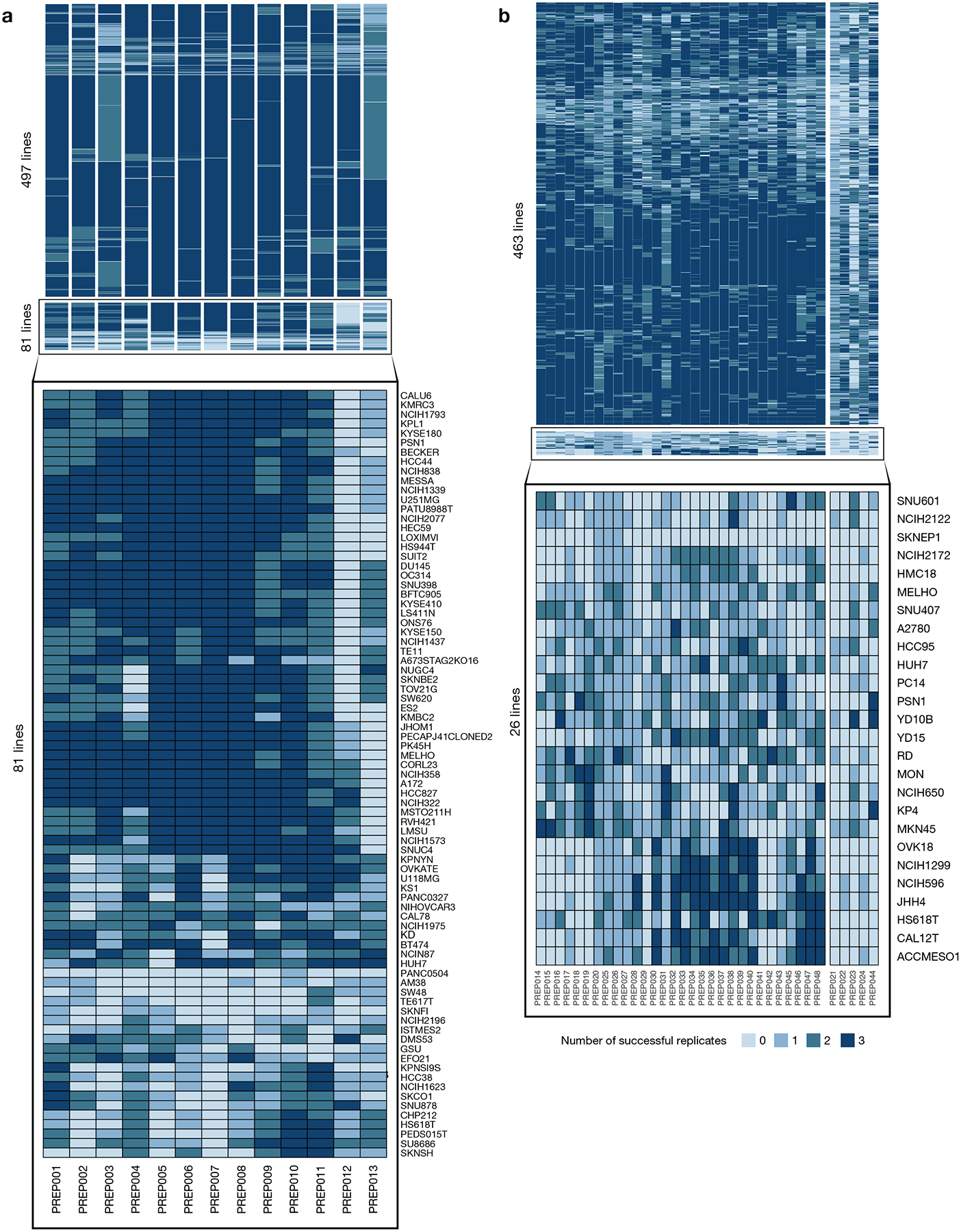
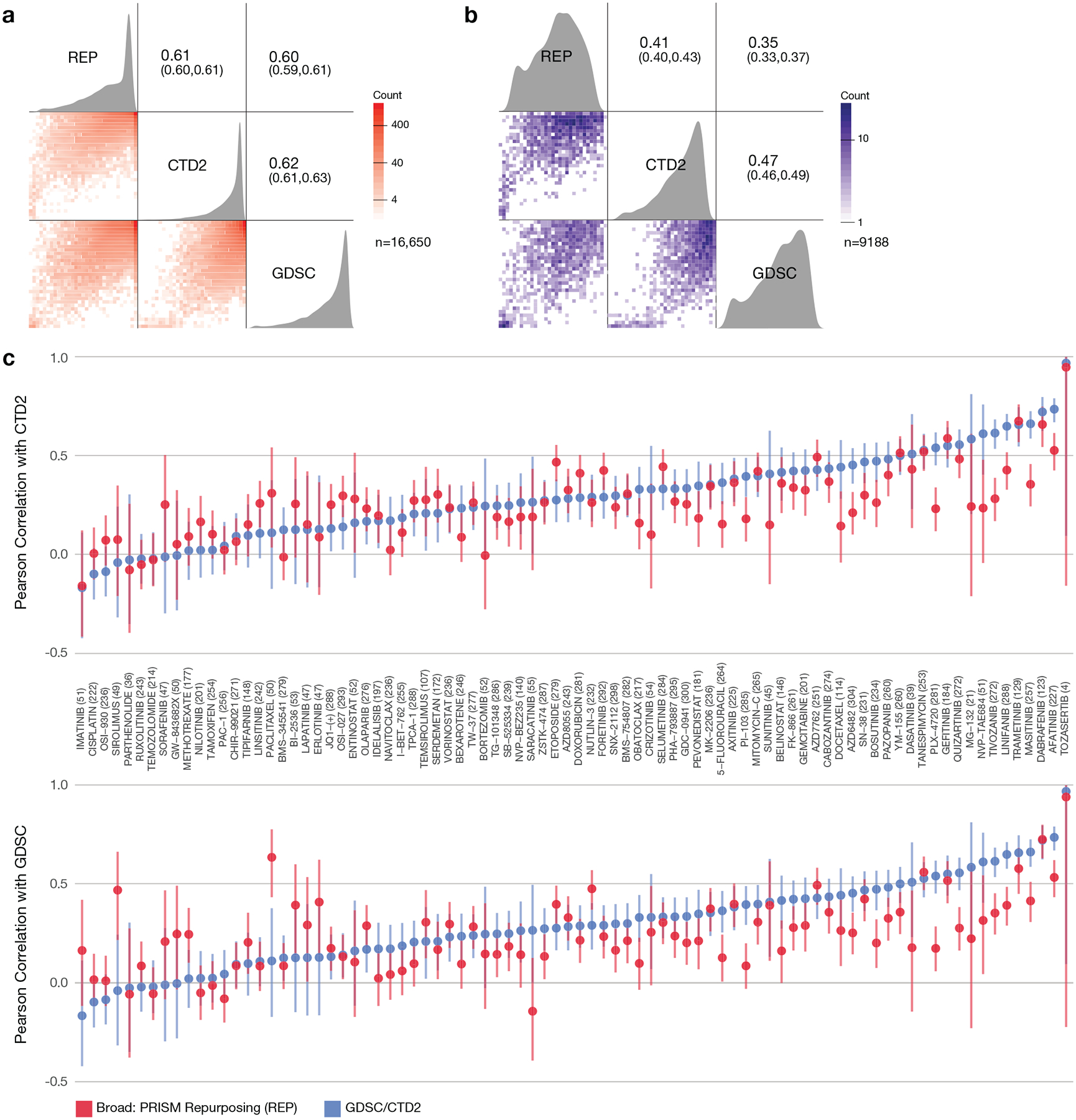
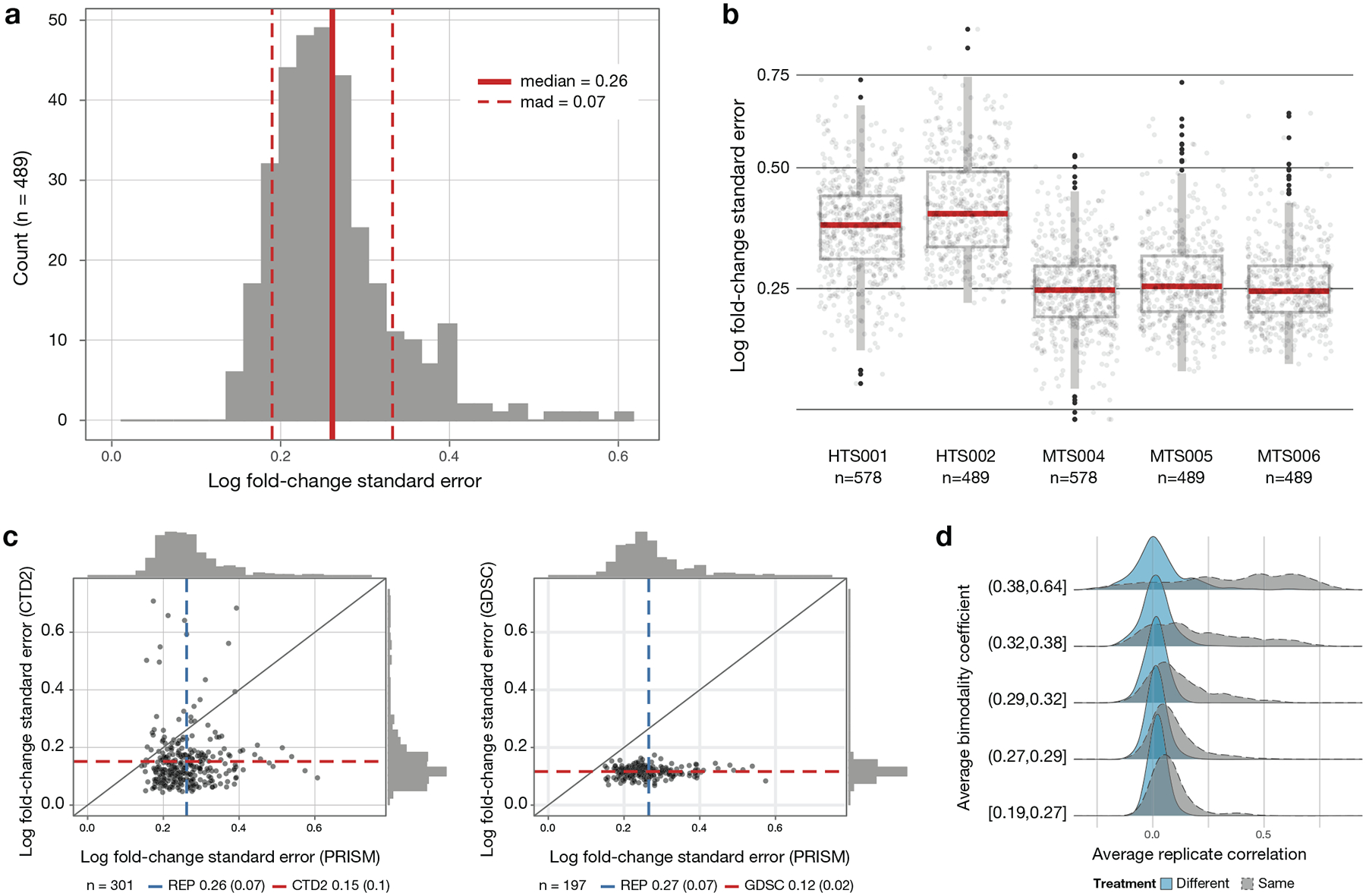
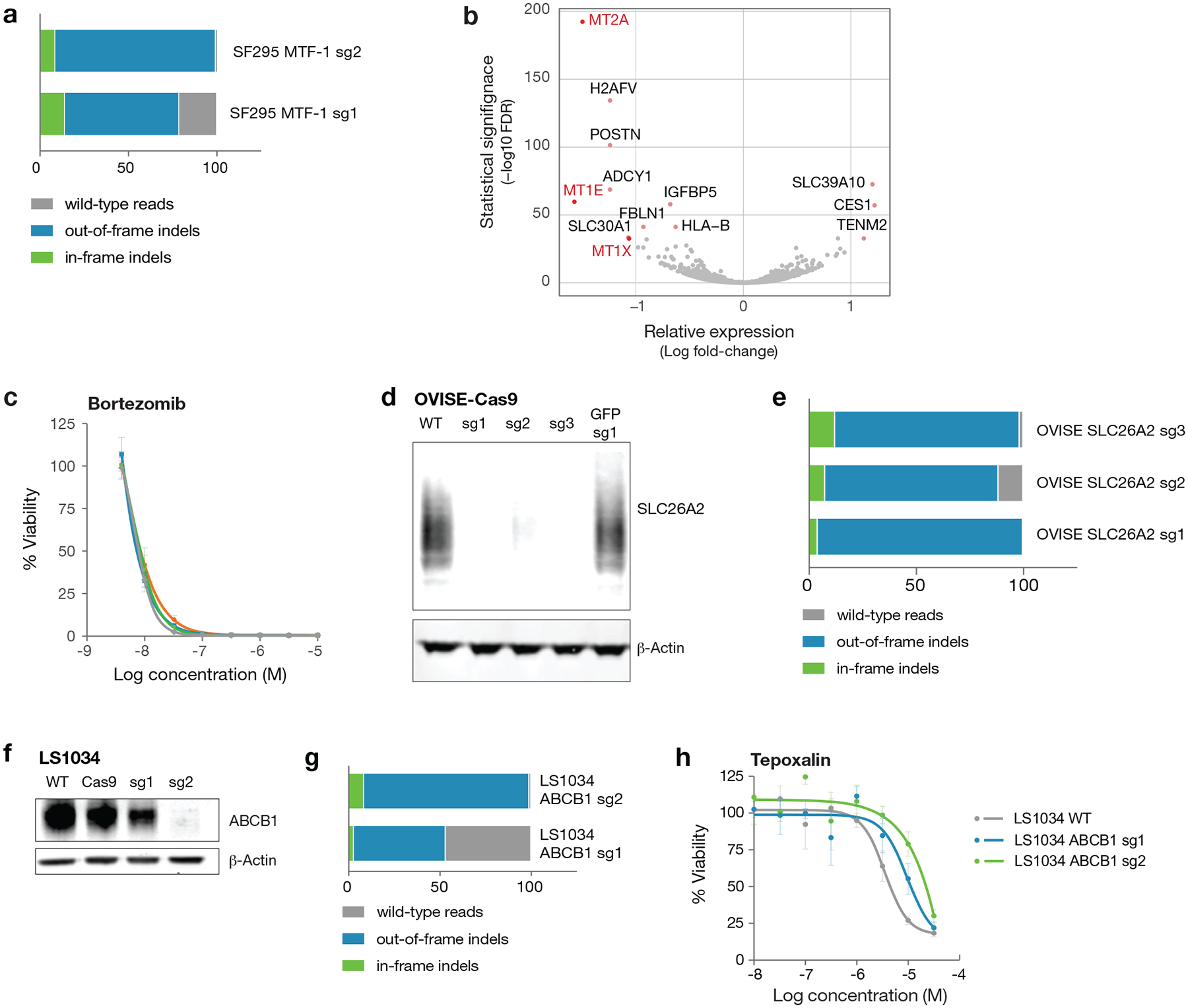
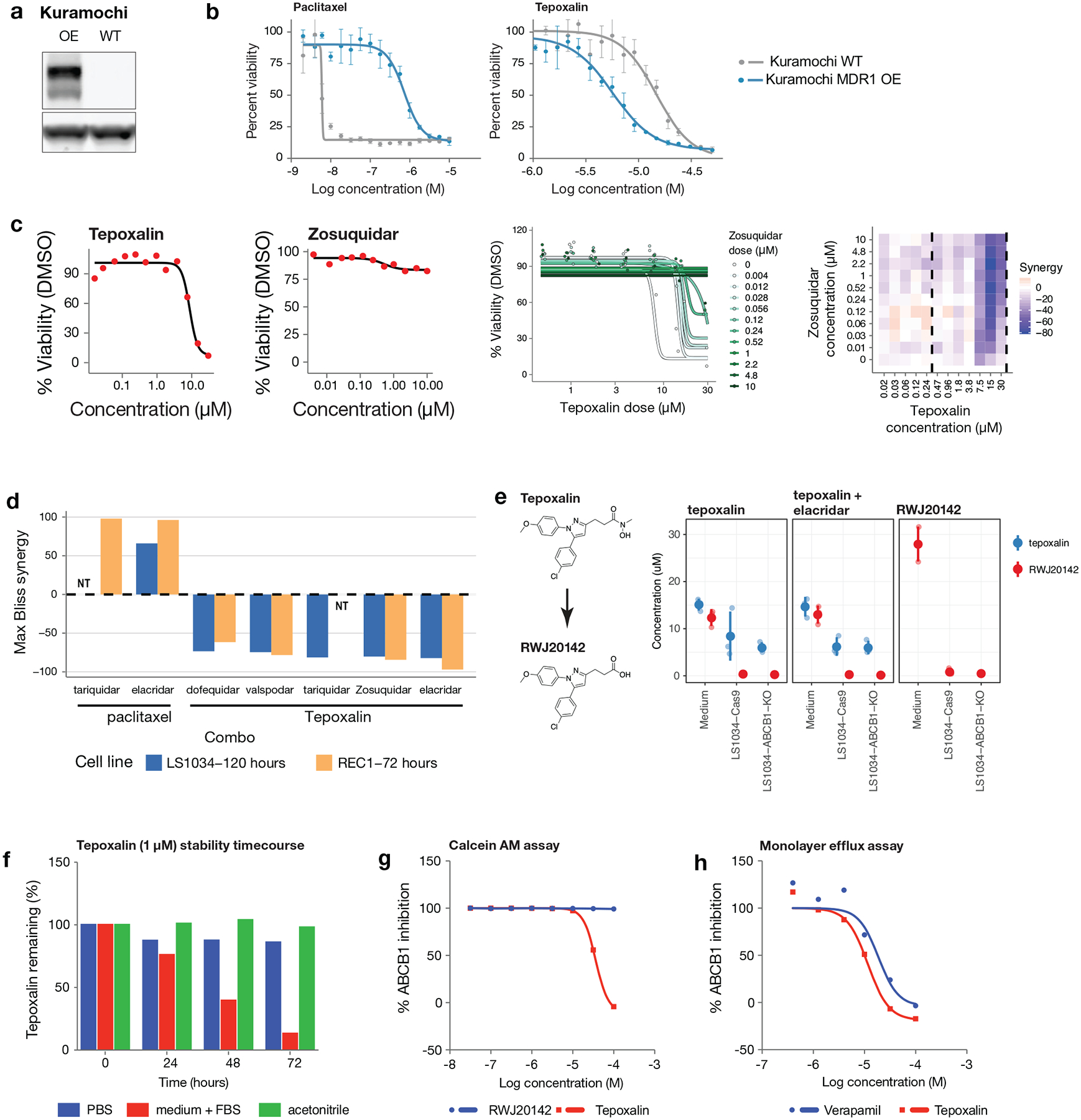
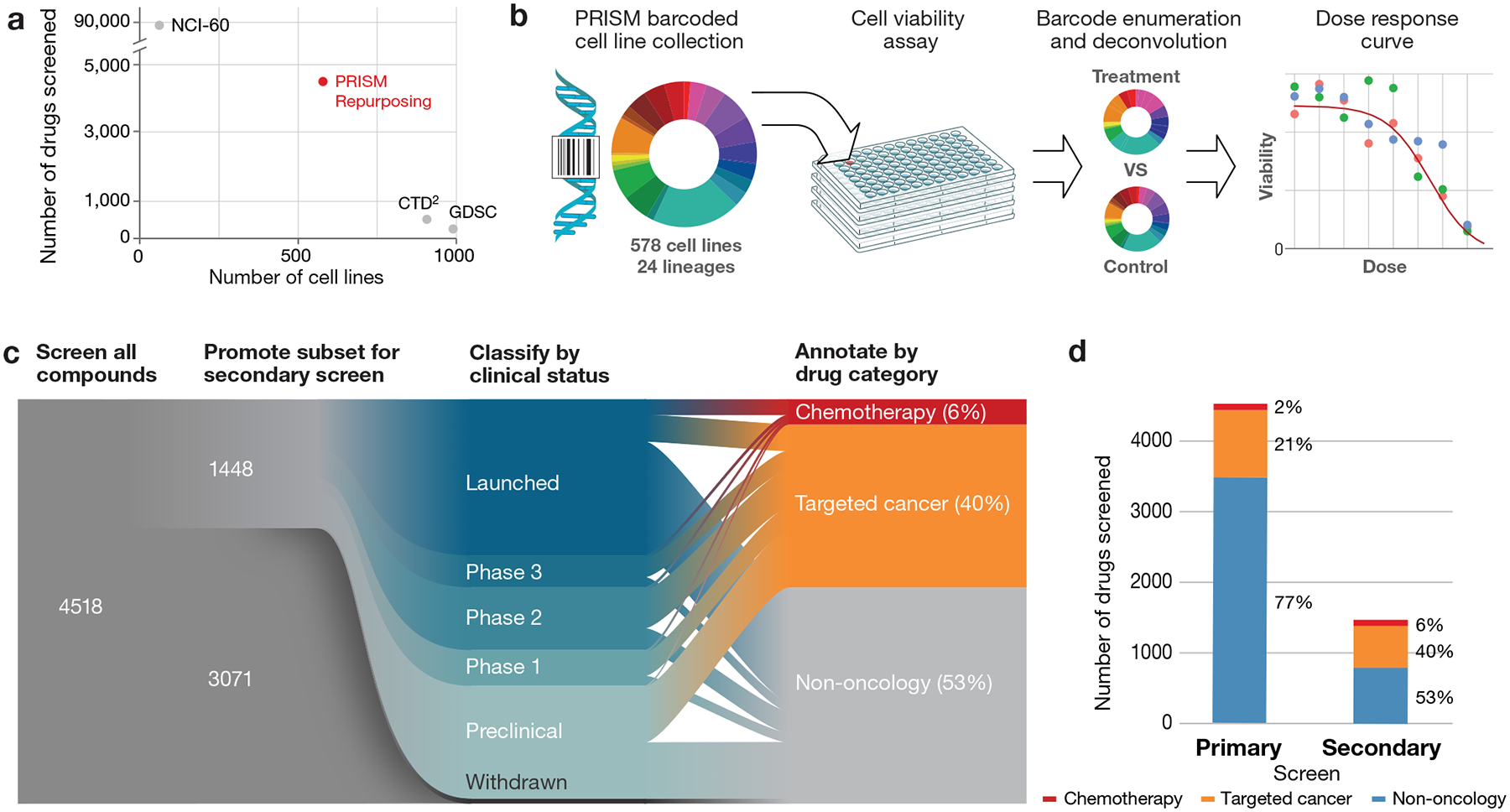
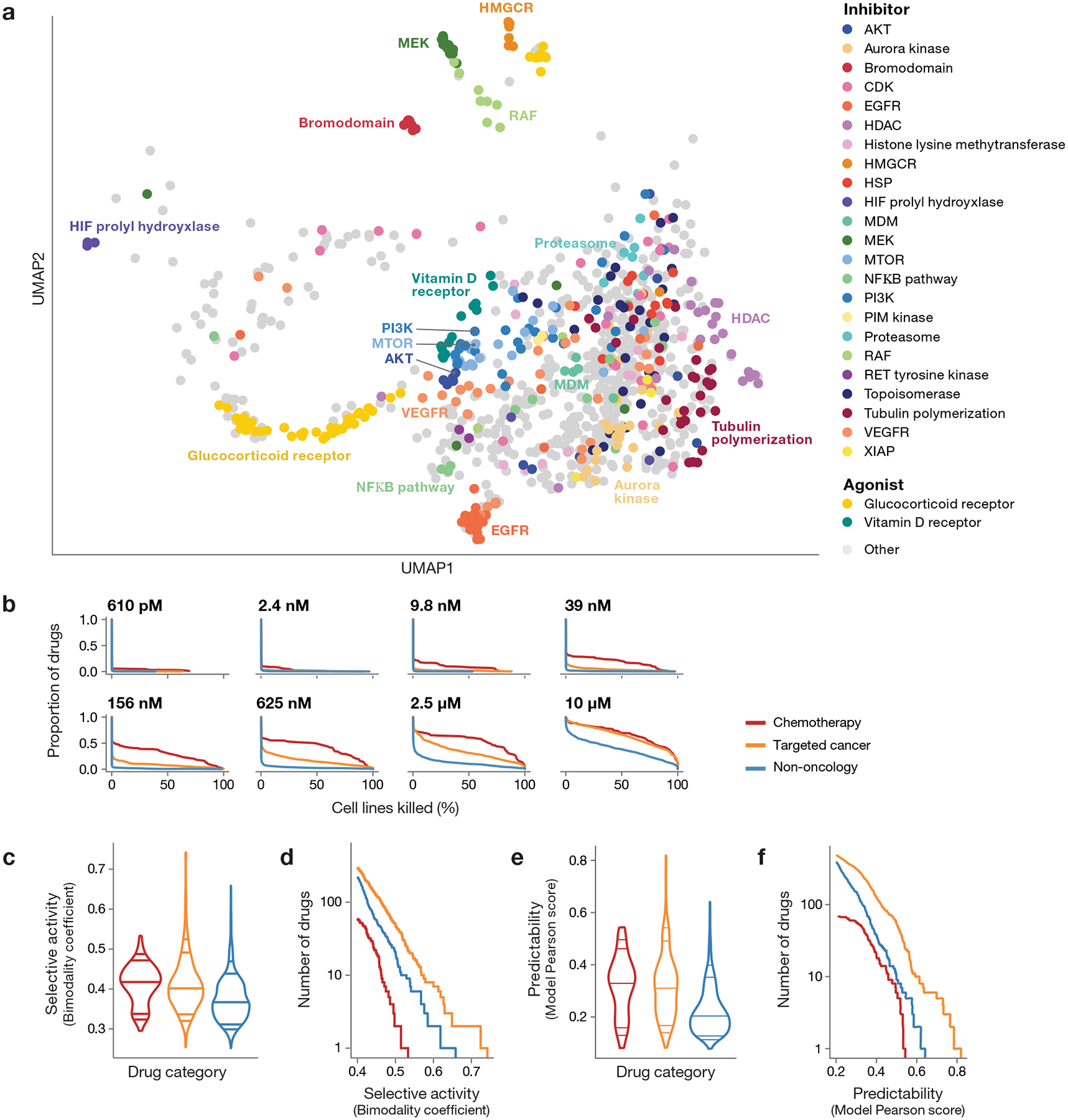
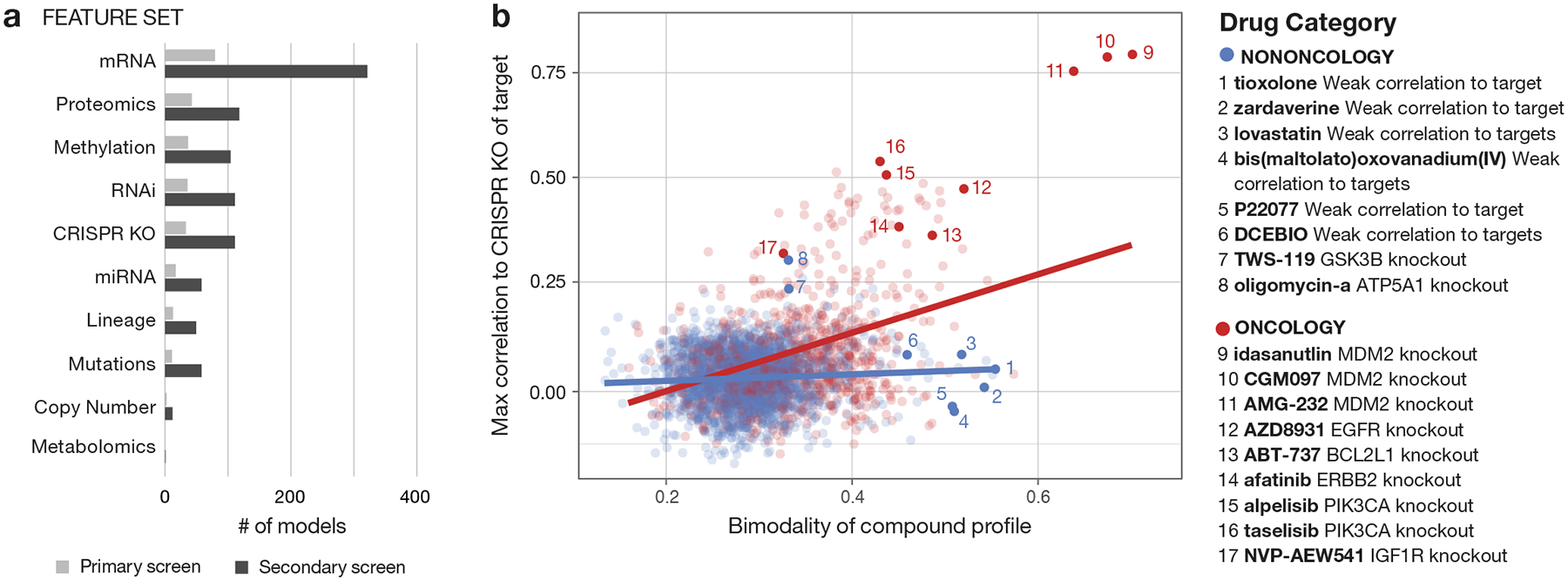
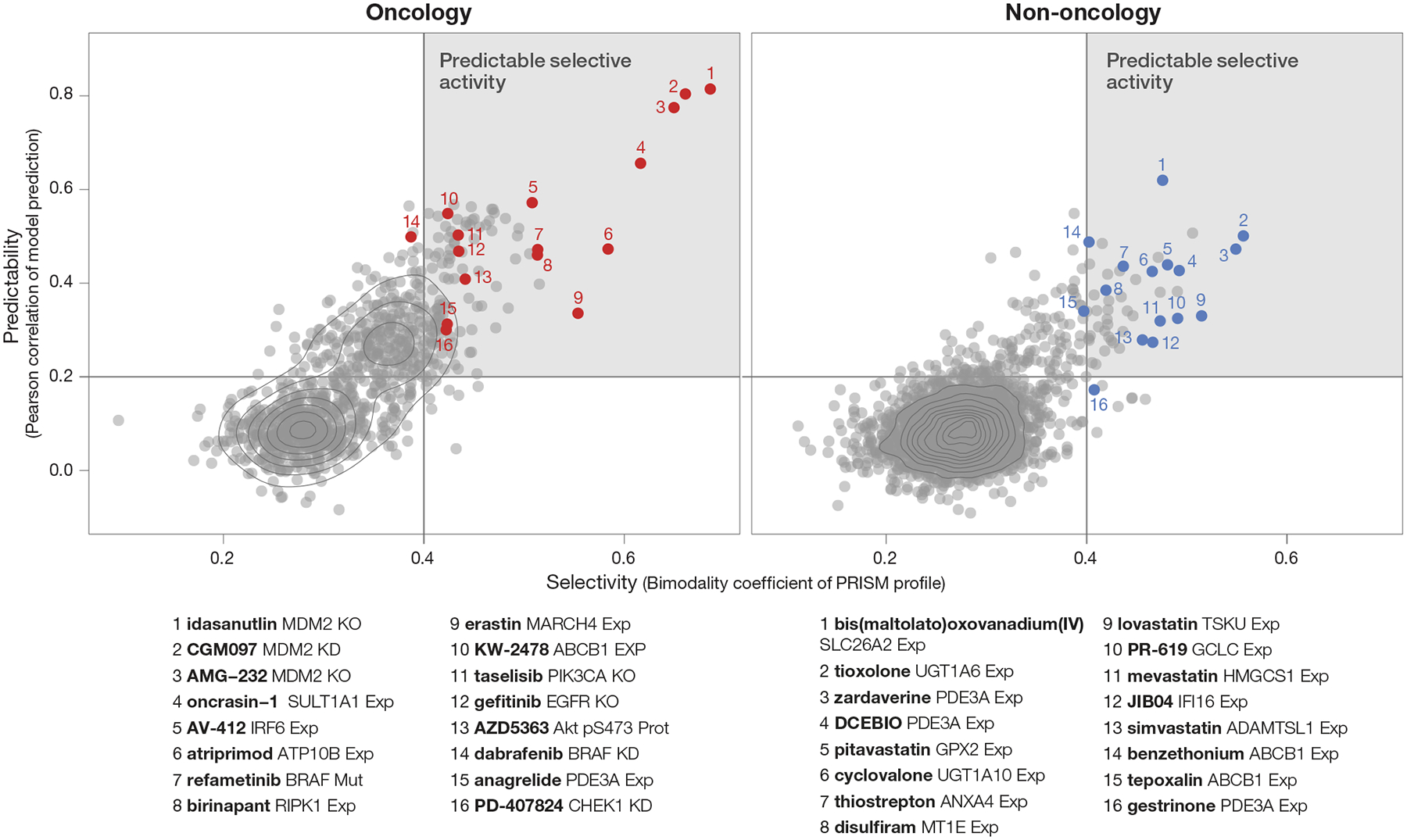
Comment in
-
Old drugs with new tricks.Nat Cancer. 2020 Feb;1(2):153-155. doi: 10.1038/s43018-020-0024-8. Nat Cancer. 2020. PMID: 35122006 No abstract available.
References
-
- Alley MC et al. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 48, 589–601 (1988). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
