

Inflammation-driven activation of JAK/STAT signaling reversibly accelerates acute myeloid leukemia in vitro
- PMID: 32614965
- PMCID: PMC7362361
- DOI: 10.1182/bloodadvances.2019001292
Inflammation-driven activation of JAK/STAT signaling reversibly accelerates acute myeloid leukemia in vitro
Abstract
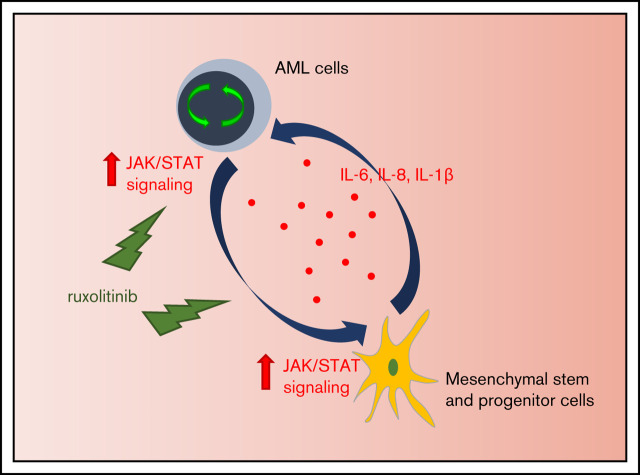
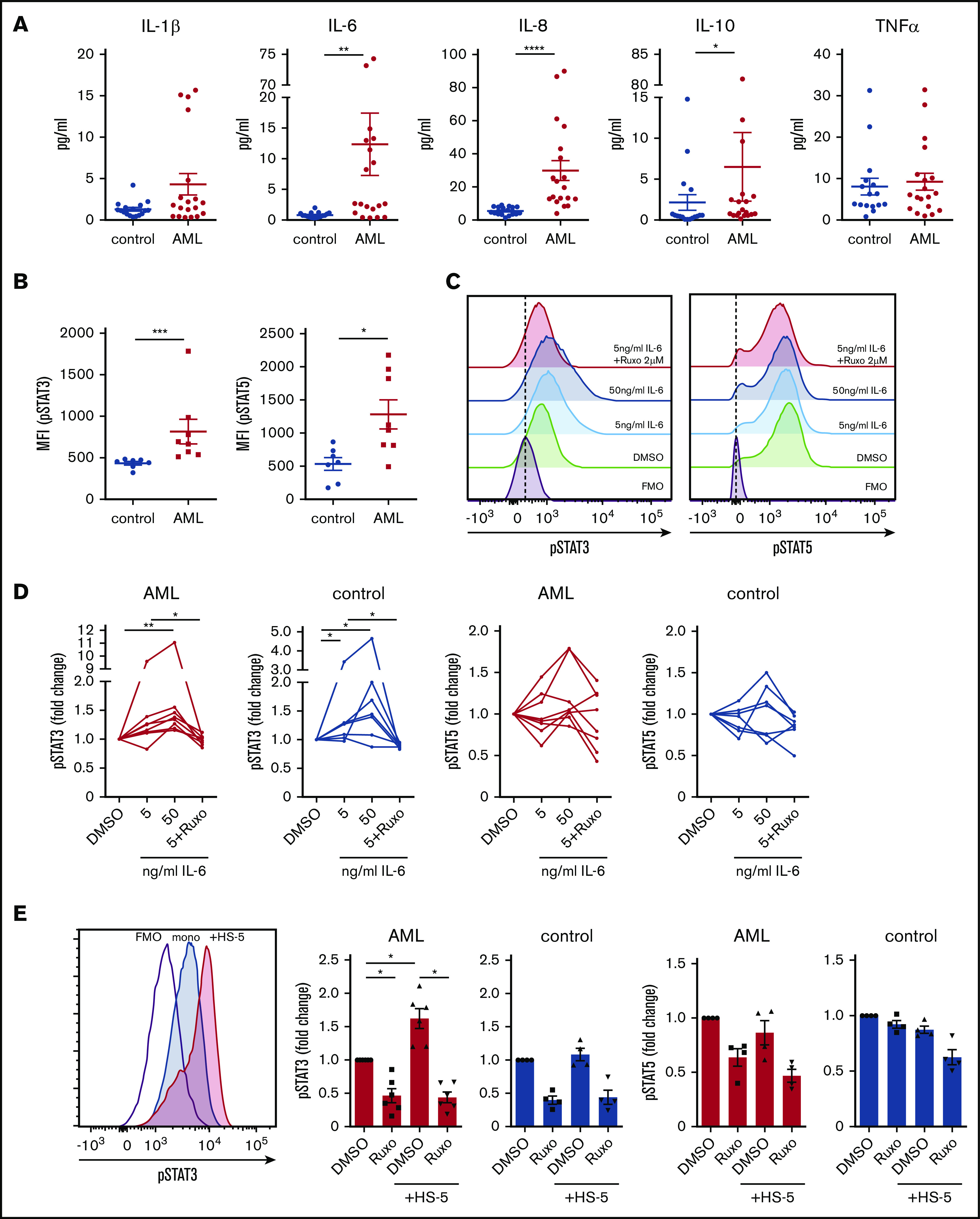
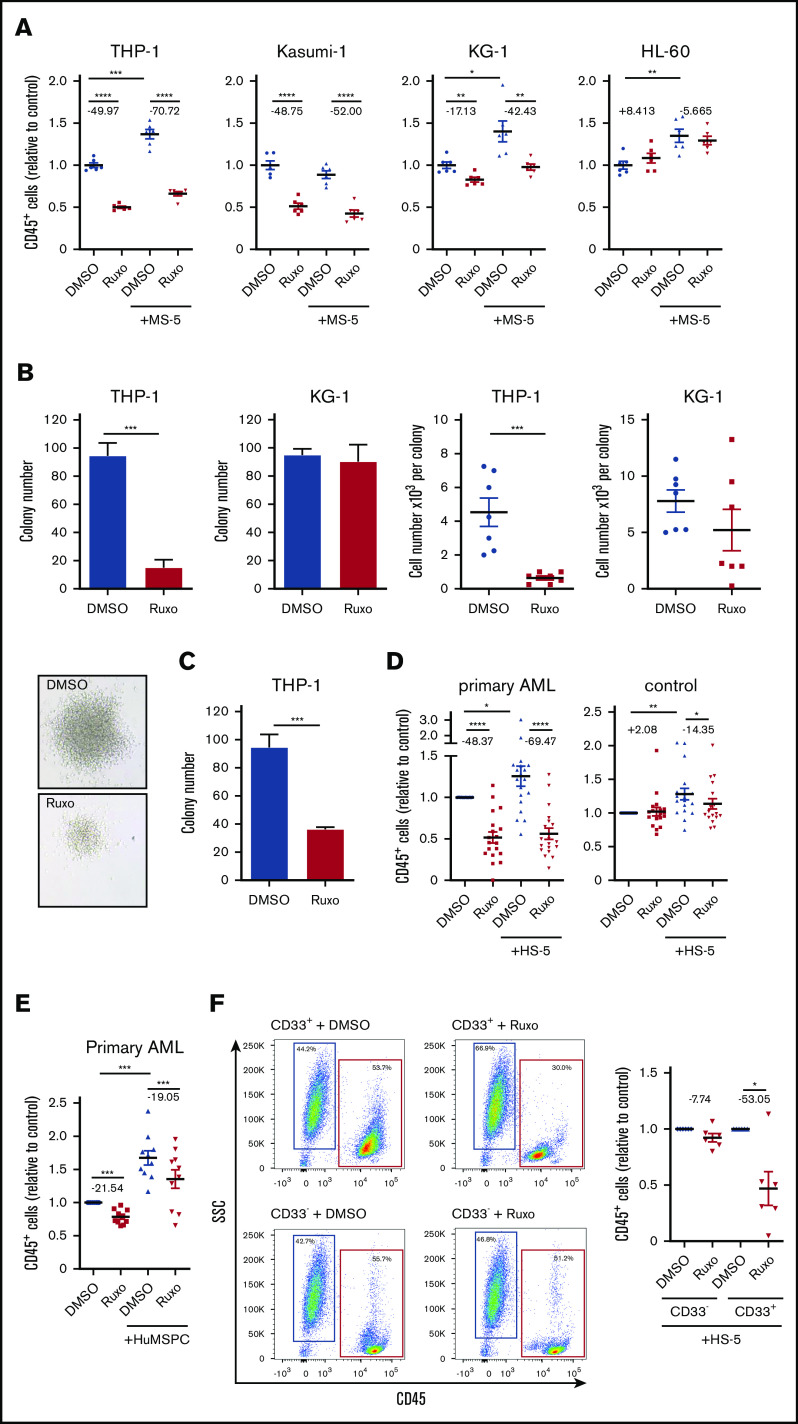
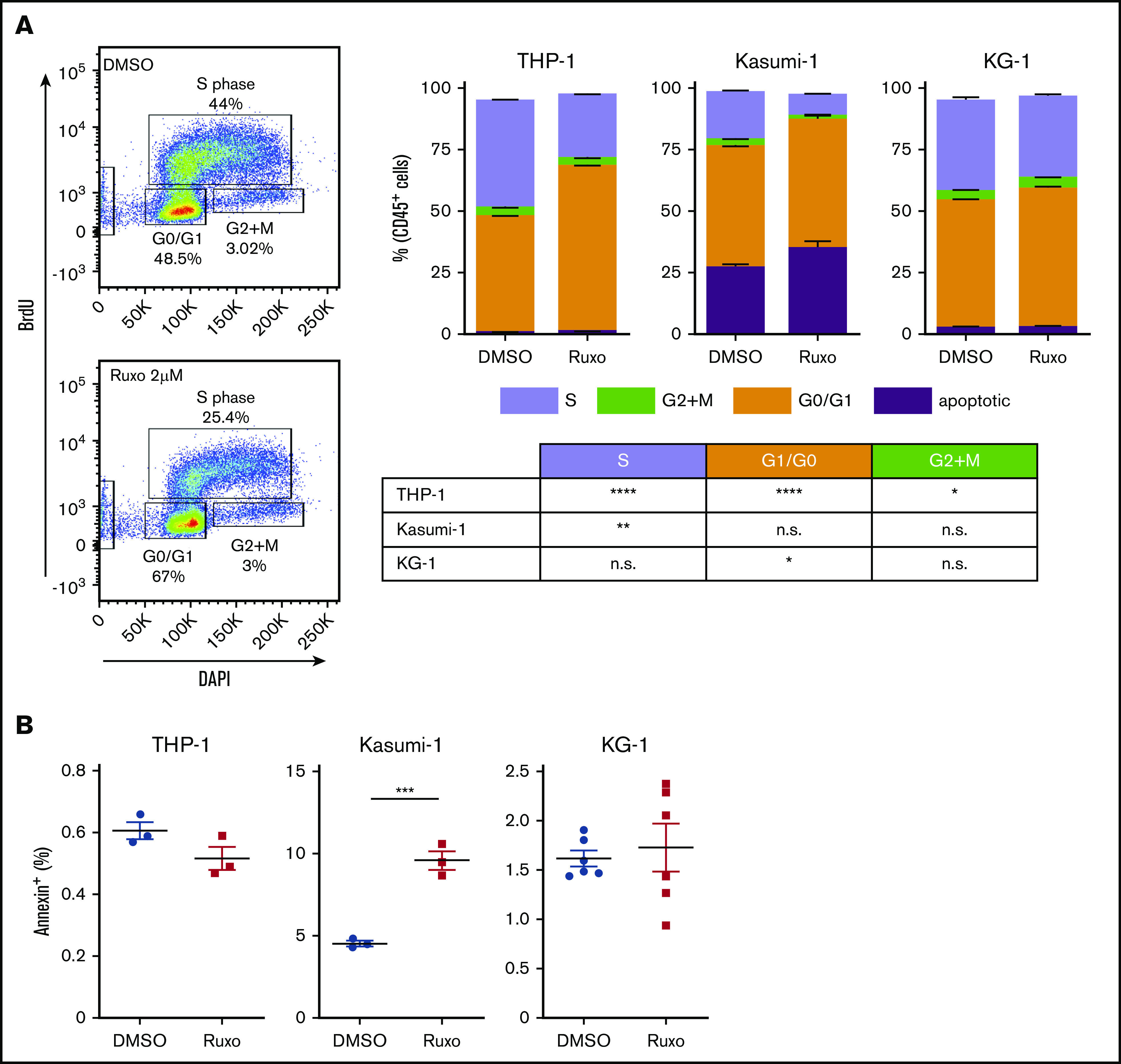
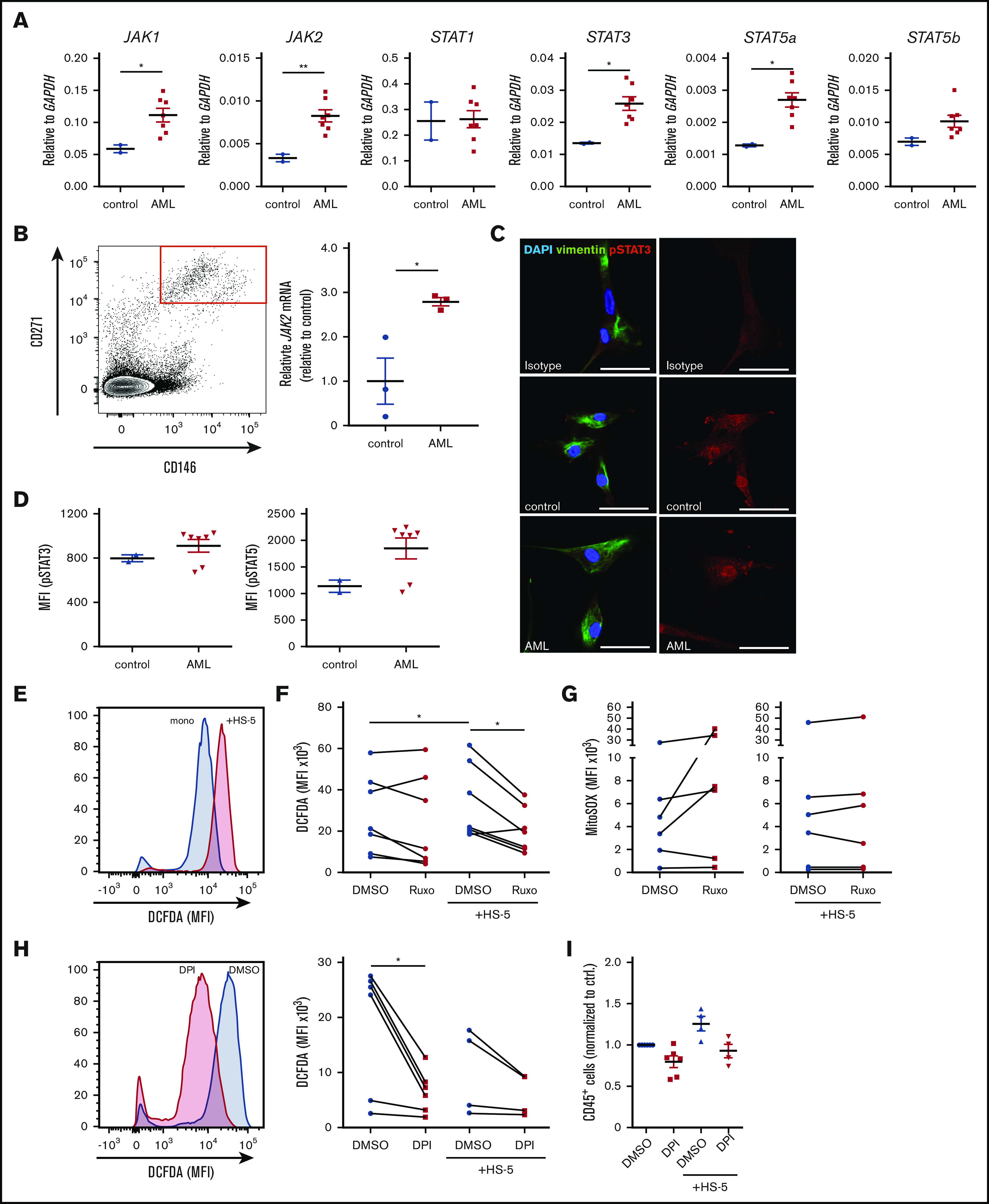
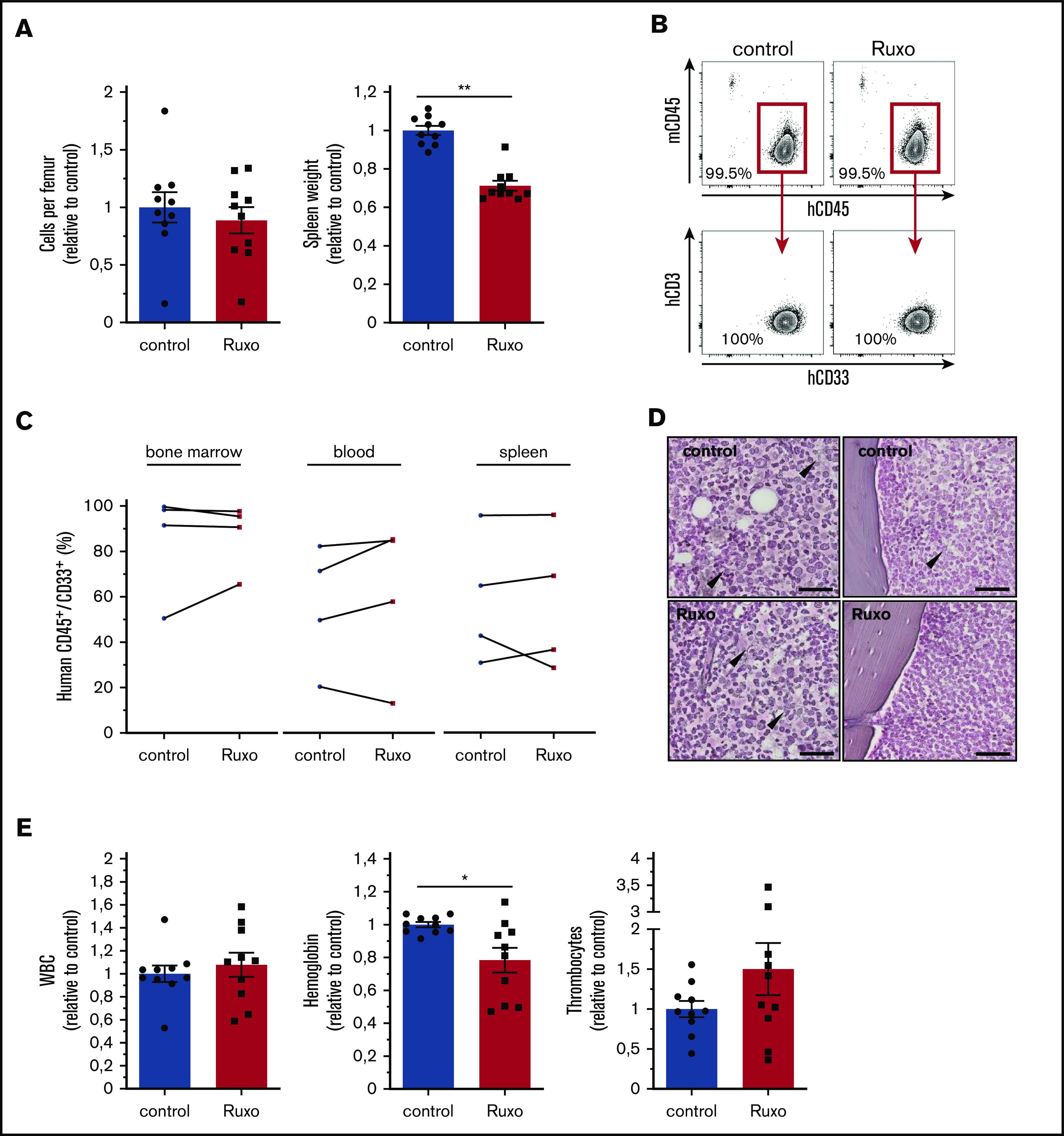
Acute myeloid leukemia (AML) is characterized by a high relapse rate and dismal long-term overall survival which is related to persistence of leukemia-initiating cells in their niche. Different animal models of myeloid malignancies reveal how neoplastic cells alter the structural and functional characteristics of the hematopoietic stem cell niche to reinforce malignancy. Understanding and disruption of the microenvironmental interactions with AML cells are a vital need. Malignant niches frequently go along with inflammatory responses, but their impact on cancerogenesis often remains unexplored. Here, we uncovered an aberrant production of inflammatory cytokines in untreated AML bone marrow that was proved to promote the proliferation of leukemia cells. This inflammatory response induced an activation of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway in AML blasts as well as bone marrow stromal cells that also fostered leukemia proliferation. Inhibition of JAK/STAT signaling using the selective JAK1/2 inhibitor ruxolitinib resulted in significant antileukemic activity in AML in vitro which is mediated through both cell-autonomous and microenvironment-mediated mechanisms. However, in a xenograft transplantation model, monotherapy with ruxolitinib did not achieve substantial antileukemic activity, possibly suggesting a complementary function of JAK1/2 inhibition in AML.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
References
-
- Medyouf H, Mossner M, Jann JC, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014;14(6):824-837. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
