

APOL1 Nephropathy: From Genetics to Clinical Applications
- PMID: 32616495
- PMCID: PMC7863644
- DOI: 10.2215/CJN.15161219
APOL1 Nephropathy: From Genetics to Clinical Applications
Abstract
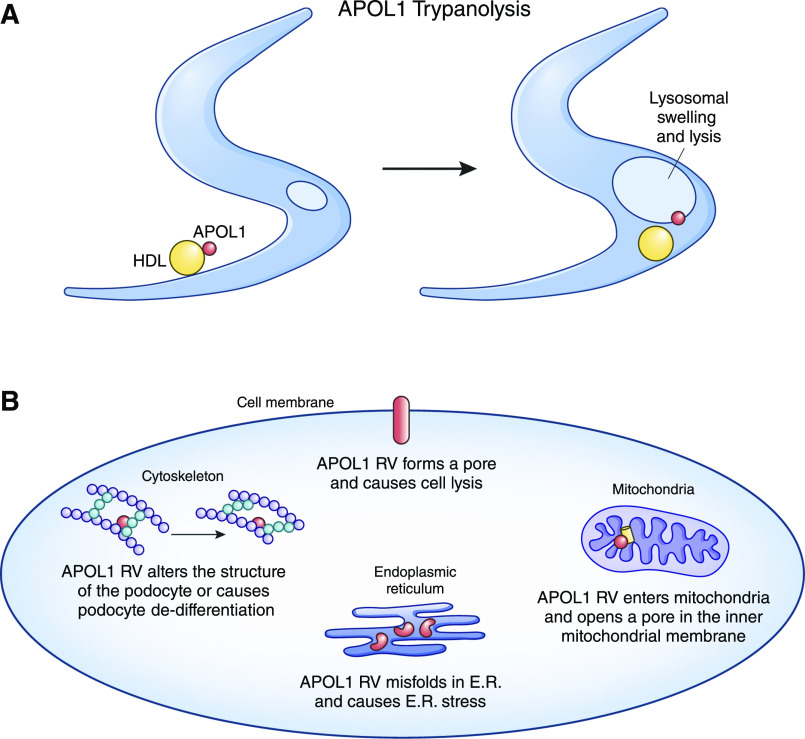
Rates of many types of severe kidney disease are much higher in Black individuals than most other ethnic groups. Much of this disparity can now be attributed to genetic variants in the apoL1 (APOL1) gene found only in individuals with recent African ancestry. These variants greatly increase rates of hypertension-associated ESKD, FSGS, HIV-associated nephropathy, and other forms of nondiabetic kidney disease. We discuss the population genetics of APOL1 risk variants and the clinical spectrum of APOL1 nephropathy. We then consider clinical issues that arise for the practicing nephrologist caring for the patient who may have APOL1 kidney disease.
Keywords: APOL1; APOL1 protein; African Americans; Apolipoprotein L1; Focal Segmental; Genetics; Glomerulosclerosis; HIV-Associated Nephropathy; Kidney Genomics Series; Population; chronic kidney disease; diabetes mellitus; genetic kidney disease; human; hypertension; kidney.
Copyright © 2021 by the American Society of Nephrology.
Figures



References
-
- Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR: Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329: 841–845, 2010. - PMC - PubMed
-
- Kruzel-Davila E, Wasser WG, Skorecki K: APOL1 nephropathy: A population genetics and evolutionary medicine detective story. Semin Nephrol 37: 490–507, 2017. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
