

Pheochromocytoma and Paraganglioma: From Epidemiology to Clinical Findings
- PMID: 32617052
- PMCID: PMC7326683
- DOI: 10.14744/SEMB.2020.18794
Pheochromocytoma and Paraganglioma: From Epidemiology to Clinical Findings
Abstract
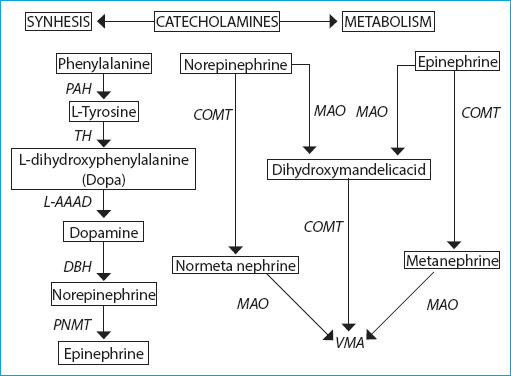
Pheochromocytomas (PCC) and paragangliomas (PGL) are rare neuroendocrine tumors. Pheochromocytomas arise from chromaffin cells in the adrenal medulla, and PGLs arise from chromaffin cells in the ganglia of the autonomic nervous system. Paragangliomas originate from sympathetic or parasympathetic ganglia in the abdomen, thorax, and pelvis. The majority of PCC and sympathetic PGL are endocrine active tumors causing clinical symptoms by secreting excess catecholamines (norepinephrine, epinephrine, dopamine) and their metabolites. The incidence of PCC and PGL ranges between 2 and 8 per million, with a prevalence between 1:2500 and 1:6500. It peaks between the 3rd and 5th decades of life, and approximately 20% of cases are pediatric patients. The prevalence among patients with hypertension in outpatient clinic ranges between 0.1-0.6% in adults and between 2-4.5% in the pediatric age group. 10-49% of these tumors is detected incidentally in imaging techniques performed for other reasons. However, 4-8% of adrenal incidentalomas are PCCs. Of these neuroendocrine tumors, 80-85% are PCCs and 15-20% are PGLs. Up to 40% of patients with PCC and PGL has disease-specific germline mutations and the situation is hereditary. Of 60% of the remaining sporadic patients, at least 1/3 has a somatic mutation in predisposing genes. 8% of the sporadic cases, 20-75% of the hereditary cases, 5% of the bilateral, adrenal cases, and 33% of the extra-adrenal cases at first presentation are metastatic. Although PCCs and PGLs have scoring systems for histological evaluation of the primary tumor, it is not possible to diagnose whether the tumor is malignant since there is no histological system approved for the biological aggressiveness of this tumor group. Metastasis is defined as the presence of chromaffin tissue in non-chromaffin organs, such as lymph nodes, liver, lungs and bone. Although most of the PCC and PGL are benign, the metastatic disease may develop in 15-17%. Metastatic disease is reported between 2-25% in PCCs and 2.4-60% in PGLs. The TNM staging system of the American Joint Committee on Cancer (AJCC) was developed to predict the prognosis, based on the specific anatomical features of the primary tumor and the occurrence of metastasis.
Keywords: Catecholamine synthesis and metabolism; paraganglioma; pheochromocytoma.
Copyright: © 2020 by The Medical Bulletin of Sisli Etfal Hospital.
Conflict of interest statement
Conflict of Interest: None declared.
Figures
References
-
- Tevosian SG, Ghayee HK. Pheochromocytomas and Paragangliomas. Endocrinol Metab Clin North Am. 2019;48:727–50. - PubMed
-
- Patel D, Phay JE, Yen TWF, Dickson PV, Wang TS, Garcia R, et al. Update on Pheochromocytoma and Paraganglioma from the SSO Endocrine/Head and Neck Disease-Site Work Group. Part 1 of 2:Advances in Pathogenesis and Diagnosis of Pheochromocytoma and Paraganglioma. Ann Surg Oncol. 2020;27:1329–37. - PMC - PubMed
-
- Schreiner F, Beuschlein F. Disease monitoring of patients with pheochromocytoma or paraganglioma by biomarkers and imaging studies. Best Pract Res Clin Endocrinol Metab. 2019 Oct 21; [Epub ahead of print], doi:10.1016/j.beem.2019.101347. - PubMed
-
- Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF, Jr Endocrine Society. Pheochromocytoma and paraganglioma:an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915–42. - PubMed
-
- Kiernan CM, Solórzano CC. Pheochromocytoma and Paraganglioma:Diagnosis, Genetics, and Treatment. Surg Oncol Clin N Am. 2016;25:119–38. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous