

A Multiplex Human Pluripotent Stem Cell Platform Defines Molecular and Functional Subclasses of Autism-Related Genes
- PMID: 32619517
- PMCID: PMC7376579
- DOI: 10.1016/j.stem.2020.06.004
A Multiplex Human Pluripotent Stem Cell Platform Defines Molecular and Functional Subclasses of Autism-Related Genes
Abstract
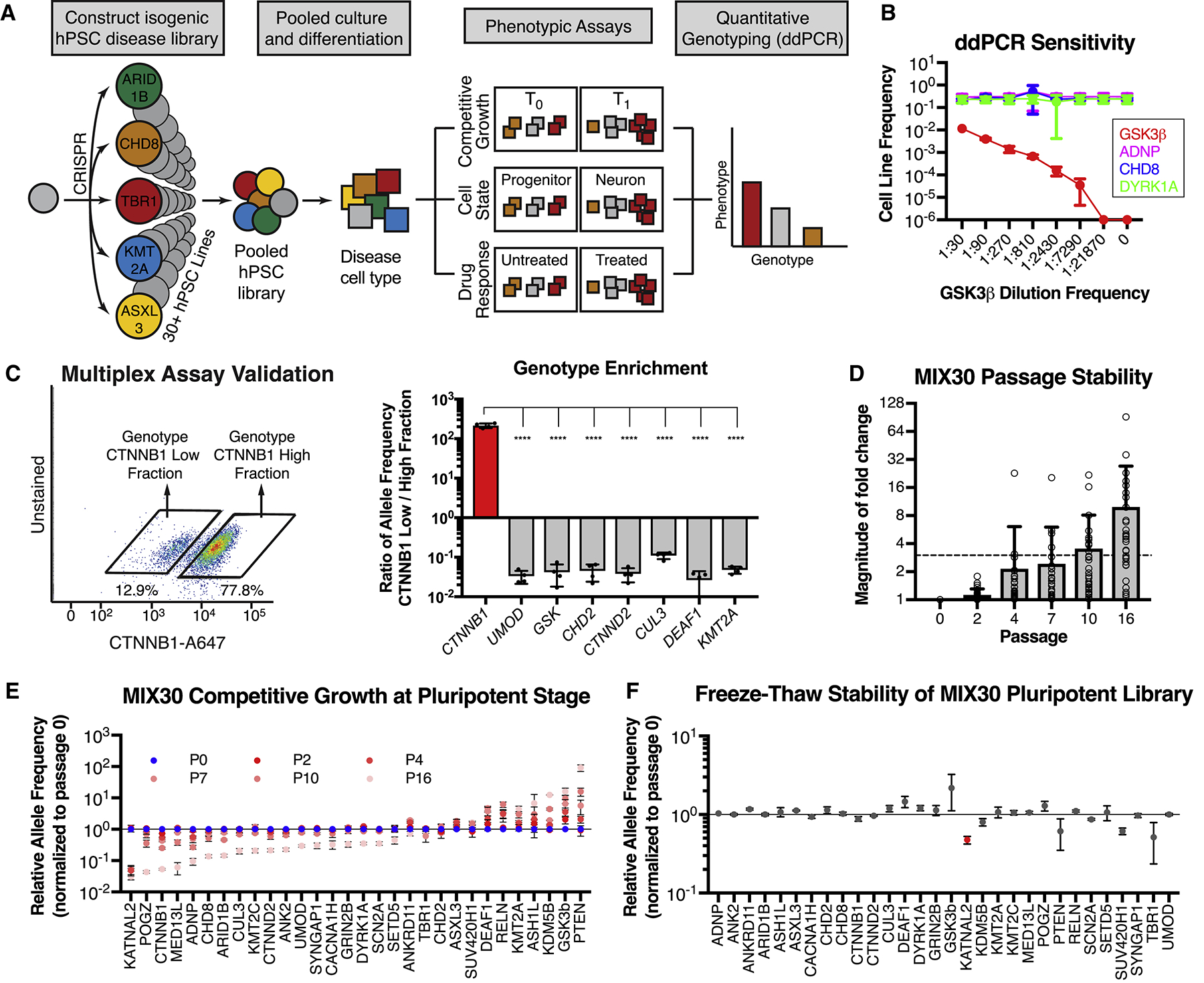
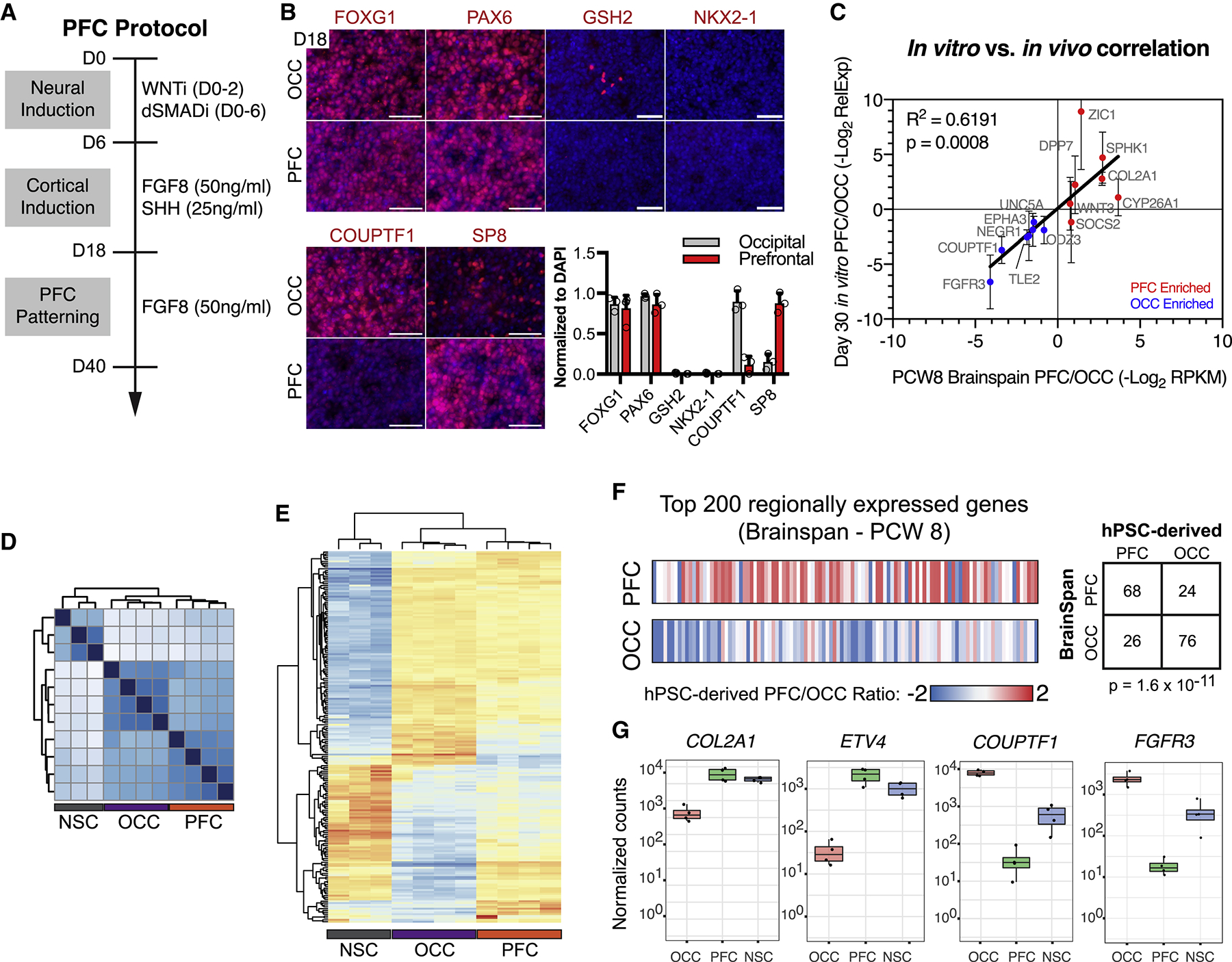
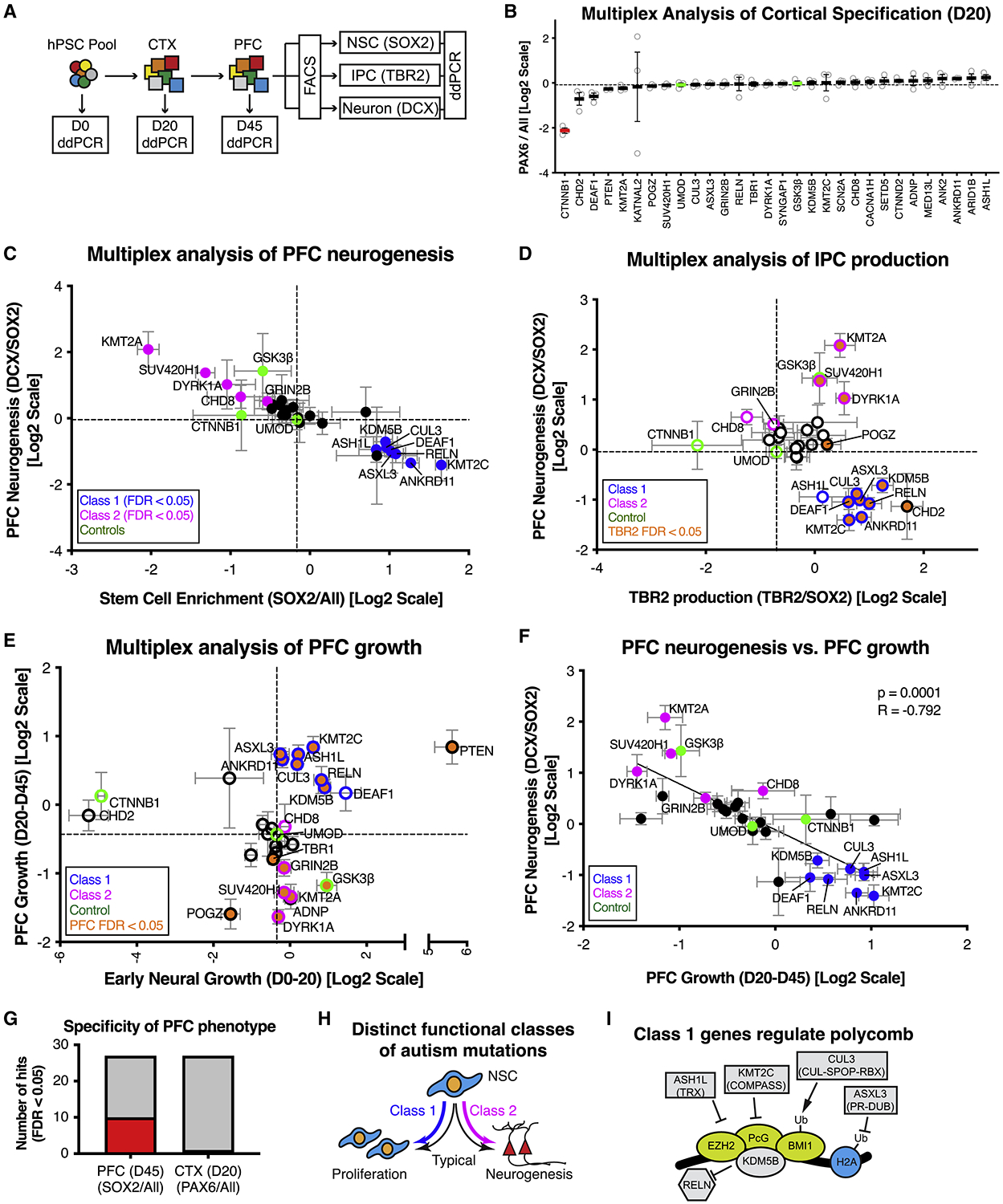
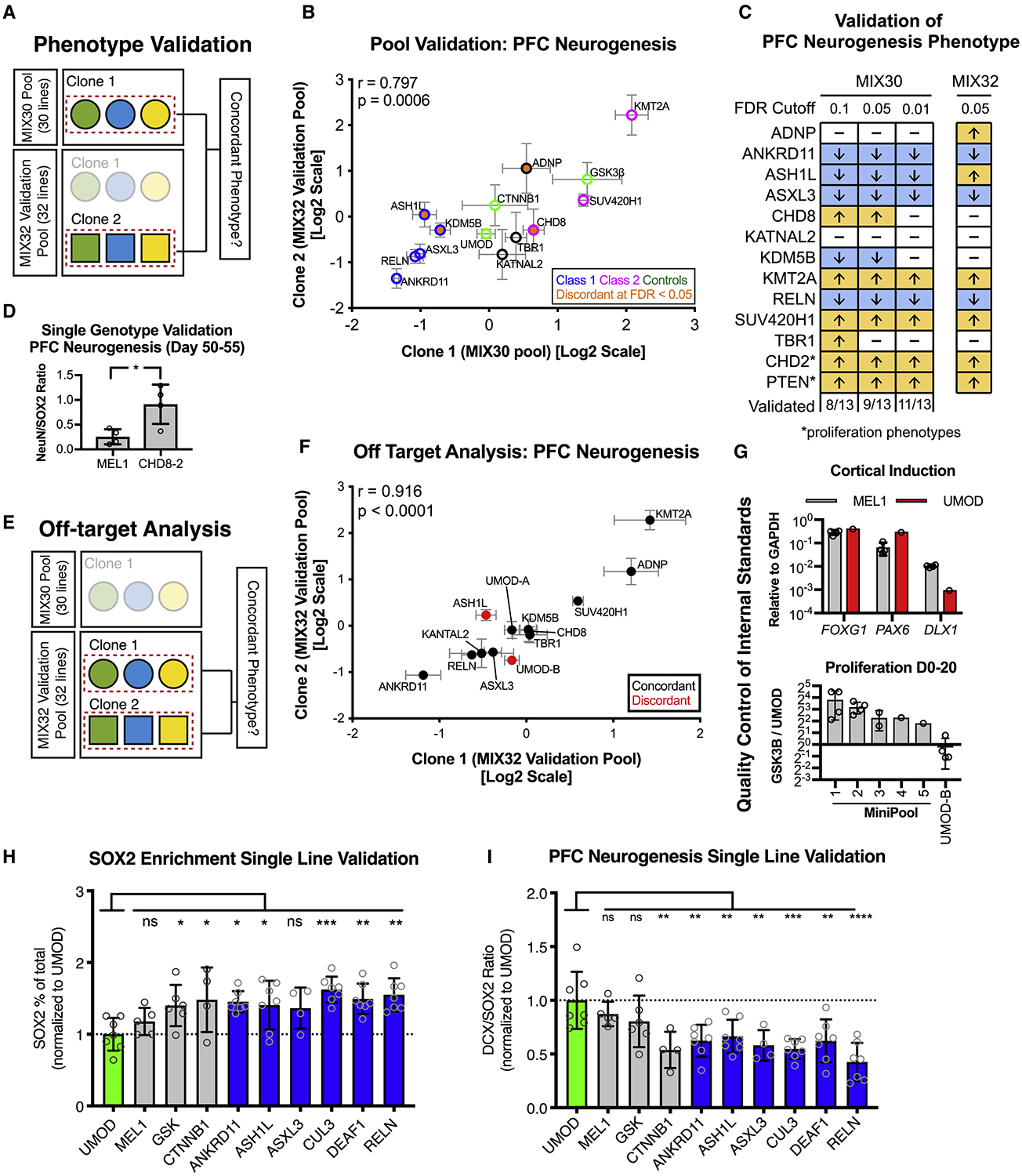
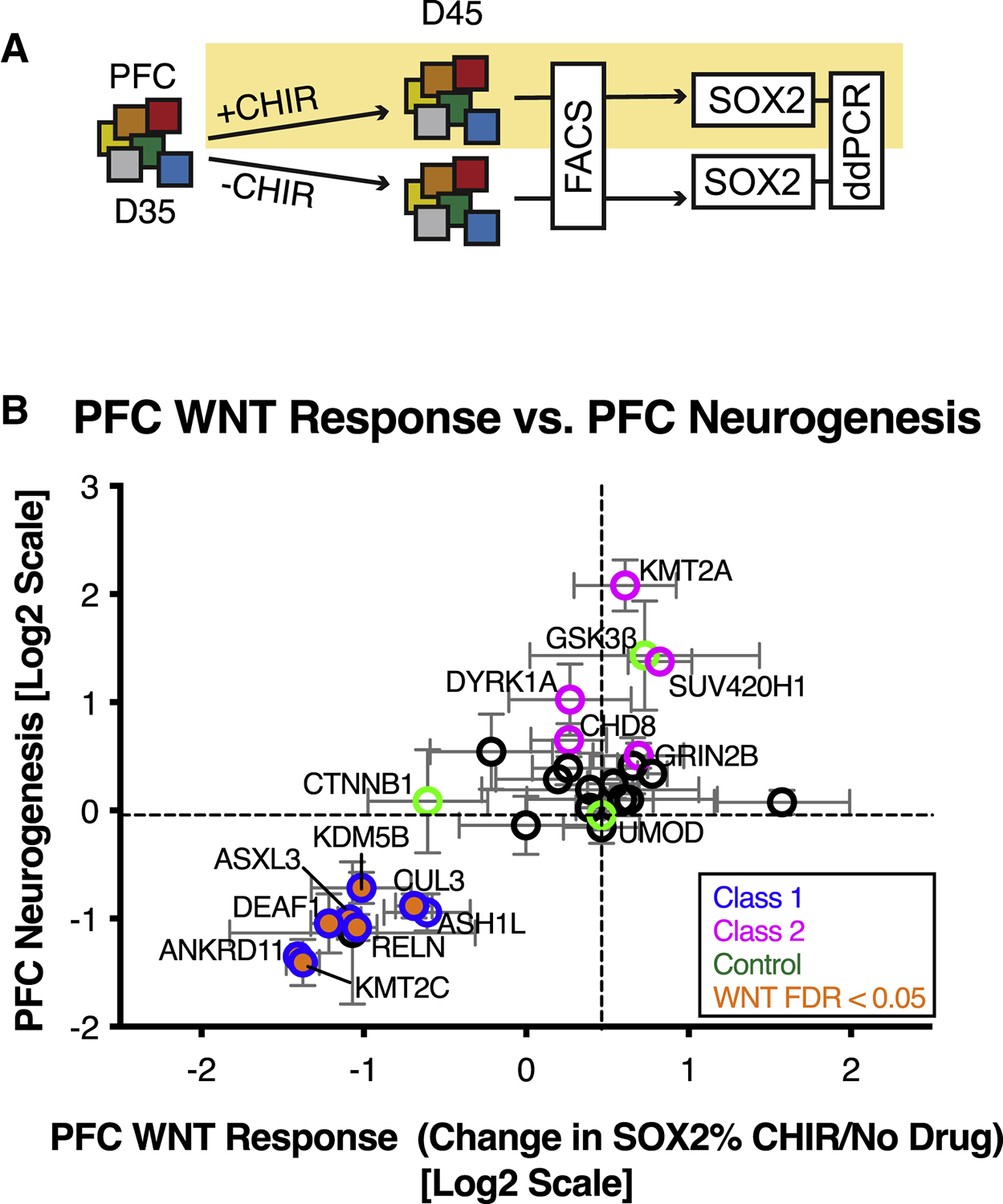
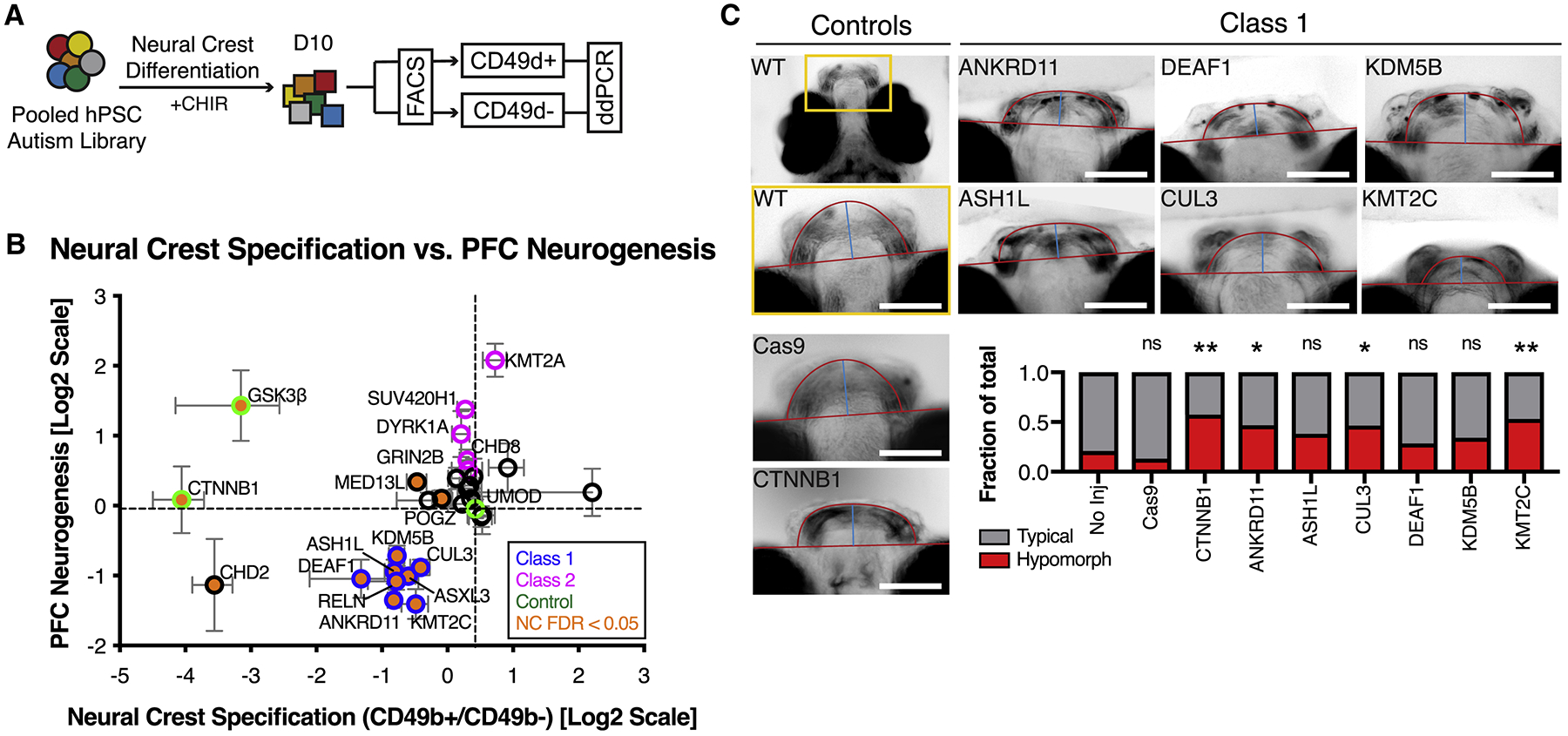
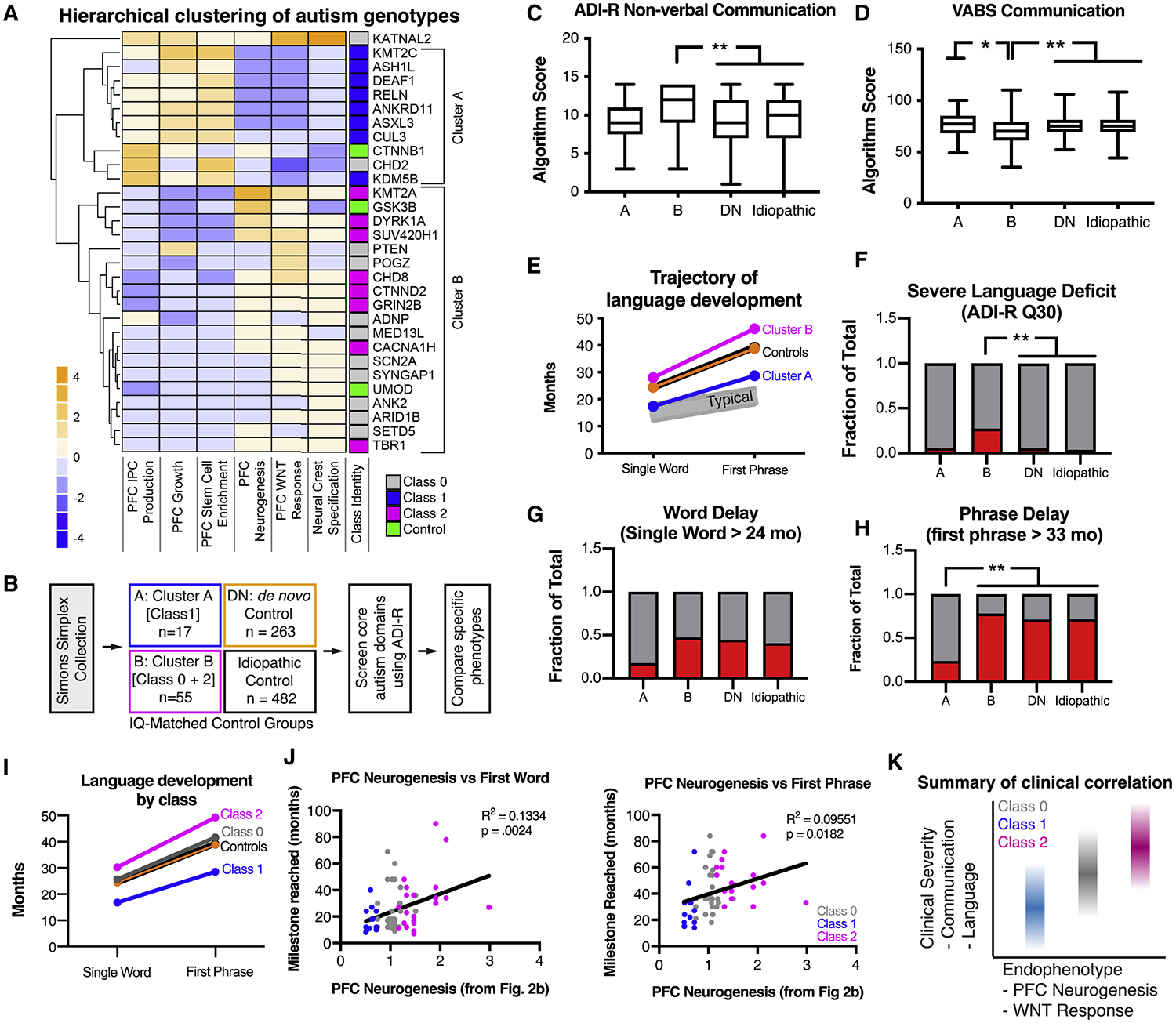
Autism is a clinically heterogeneous neurodevelopmental disorder characterized by impaired social interactions, restricted interests, and repetitive behaviors. Despite significant advances in the genetics of autism, understanding how genetic changes perturb brain development and affect clinical symptoms remains elusive. Here, we present a multiplex human pluripotent stem cell (hPSC) platform, in which 30 isogenic disease lines are pooled in a single dish and differentiated into prefrontal cortex (PFC) lineages to efficiently test early-developmental hypotheses of autism. We define subgroups of autism mutations that perturb PFC neurogenesis and are correlated to abnormal WNT/βcatenin responses. Class 1 mutations (8 of 27) inhibit while class 2 mutations (5 of 27) enhance PFC neurogenesis. Remarkably, autism patient data reveal that individuals carrying subclass-specific mutations differ clinically in their corresponding language acquisition profiles. Our study provides a framework to disentangle genetic heterogeneity associated with autism and points toward converging molecular and developmental pathways of diverse autism-associated mutations.
Keywords: autism; genetics; human pluripotent stem cells; neural development; prefrontal cortex.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests G.Y.C. and L.S. are listed as inventors of a related patent application filed by the Memorial Sloan Kettering Cancer Center. L.S. is a co-founder and paid consultant of BlueRock Therapeutics.
Figures
Comment in
-
One for All: A Pooled Approach to Classify Functional Impacts of Multiple Mutations.Cell Stem Cell. 2020 Jul 2;27(1):1-3. doi: 10.1016/j.stem.2020.06.016. Cell Stem Cell. 2020. PMID: 32619508
Similar articles
-
Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile X syndrome.Brain. 2017 Mar 1;140(3):582-598. doi: 10.1093/brain/aww357. Brain. 2017. PMID: 28137726 Free PMC article.
-
The genetics of autism.Pediatrics. 2004 May;113(5):e472-86. doi: 10.1542/peds.113.5.e472. Pediatrics. 2004. PMID: 15121991 Review.
-
Cellular and molecular characterization of multiplex autism in human induced pluripotent stem cell-derived neurons.Mol Autism. 2019 Dec 30;10:51. doi: 10.1186/s13229-019-0306-0. eCollection 2019. Mol Autism. 2019. PMID: 31893020 Free PMC article.
-
Atypical Neurogenesis in Induced Pluripotent Stem Cells From Autistic Individuals.Biol Psychiatry. 2021 Mar 1;89(5):486-496. doi: 10.1016/j.biopsych.2020.06.014. Epub 2020 Jun 23. Biol Psychiatry. 2021. PMID: 32826066 Free PMC article.
-
Psychiatry in a Dish: Stem Cells and Brain Organoids Modeling Autism Spectrum Disorders.Biol Psychiatry. 2018 Apr 1;83(7):558-568. doi: 10.1016/j.biopsych.2017.11.011. Epub 2017 Nov 16. Biol Psychiatry. 2018. PMID: 29295738 Review.
Cited by
-
Reaching into the toolbox: Stem cell models to study neuropsychiatric disorders.Stem Cell Reports. 2022 Feb 8;17(2):187-210. doi: 10.1016/j.stemcr.2021.12.015. Epub 2022 Jan 20. Stem Cell Reports. 2022. PMID: 35063127 Free PMC article. Review.
-
Humanized brain organoids-on-chip integrated with sensors for screening neuronal activity and neurotoxicity.Mikrochim Acta. 2024 Jan 3;191(1):71. doi: 10.1007/s00604-023-06165-4. Mikrochim Acta. 2024. PMID: 38168828 Review.
-
INTERSPECIES ORGANOIDS REVEAL HUMAN-SPECIFIC MOLECULAR FEATURES OF DOPAMINERGIC NEURON DEVELOPMENT AND VULNERABILITY.bioRxiv [Preprint]. 2024 Nov 15:2024.11.14.623592. doi: 10.1101/2024.11.14.623592. bioRxiv. 2024. PMID: 39605599 Free PMC article. Preprint.
-
Intersectionality and genetic ancestry: New methods to solve old problems.EBioMedicine. 2022 Jun;80:104049. doi: 10.1016/j.ebiom.2022.104049. Epub 2022 May 10. EBioMedicine. 2022. PMID: 35561454 Free PMC article. No abstract available.
-
Cerebral Organoids as an Experimental Platform for Human Neurogenomics.Cells. 2022 Sep 8;11(18):2803. doi: 10.3390/cells11182803. Cells. 2022. PMID: 36139380 Free PMC article. Review.
References
-
- Balasubramanian M, Willoughby J, Fry AE, Weber A, Firth HV, Deshpande C, Berg JN, Chandler K, Metcalfe KA, Lam W, et al. (2017). Delineating the phenotypic spectrum of Bainbridge-Ropers syndrome: 12 new patients with de novo, heterozygous, loss-of-function mutations in ASXL3 and review of published literature. J Med Genet 54, 537–543. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
