

Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection
- PMID: 32619549
- PMCID: PMC7264033
- DOI: 10.1016/j.cels.2020.05.012
Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection
Abstract
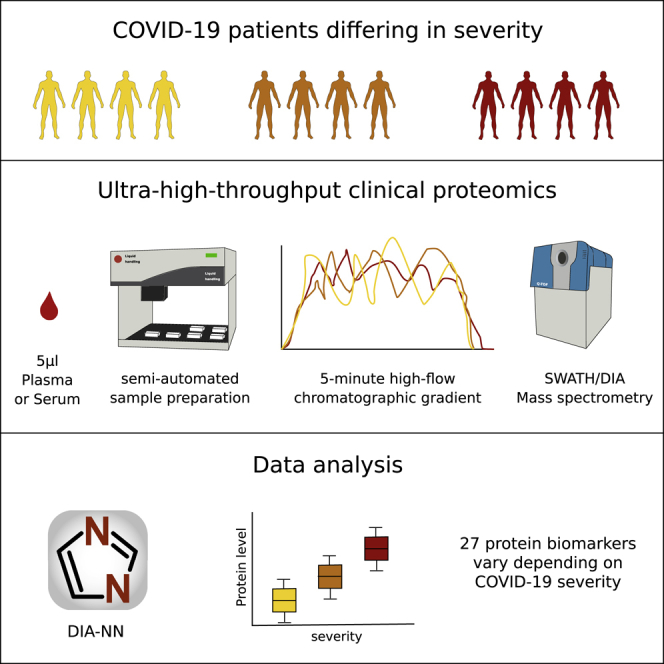
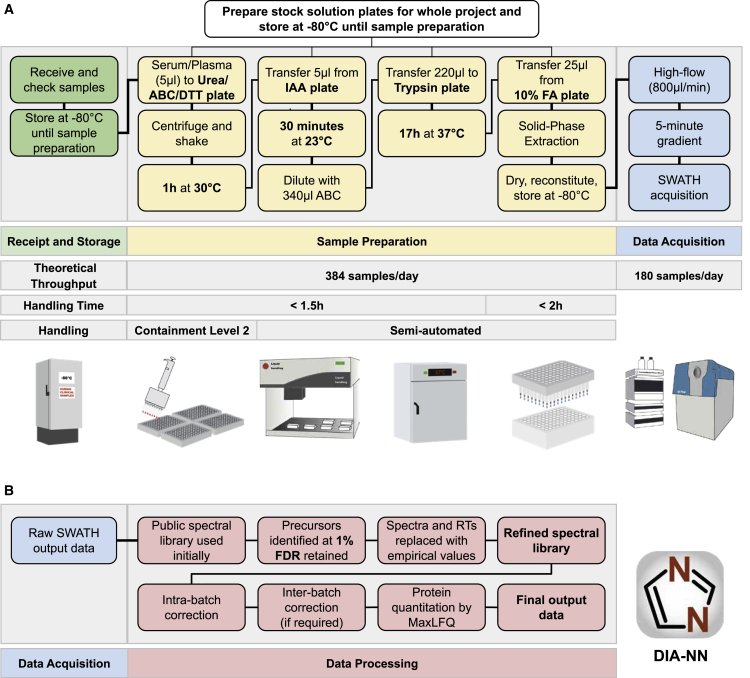
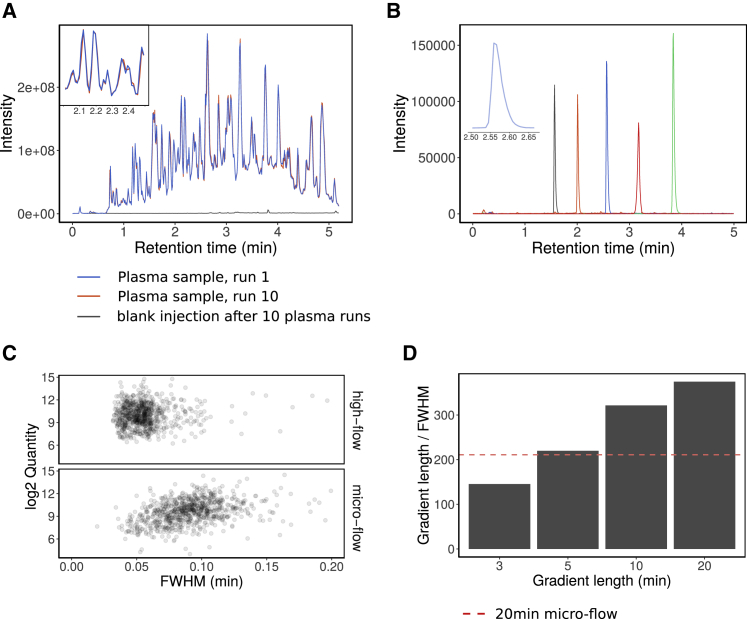
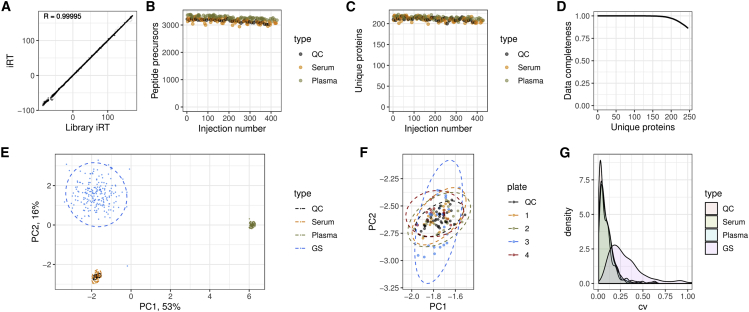
The COVID-19 pandemic is an unprecedented global challenge, and point-of-care diagnostic classifiers are urgently required. Here, we present a platform for ultra-high-throughput serum and plasma proteomics that builds on ISO13485 standardization to facilitate simple implementation in regulated clinical laboratories. Our low-cost workflow handles up to 180 samples per day, enables high precision quantification, and reduces batch effects for large-scale and longitudinal studies. We use our platform on samples collected from a cohort of early hospitalized cases of the SARS-CoV-2 pandemic and identify 27 potential biomarkers that are differentially expressed depending on the WHO severity grade of COVID-19. They include complement factors, the coagulation system, inflammation modulators, and pro-inflammatory factors upstream and downstream of interleukin 6. All protocols and software for implementing our approach are freely available. In total, this work supports the development of routine proteomic assays to aid clinical decision making and generate hypotheses about potential COVID-19 therapeutic targets.
Keywords: COVID-19 infection; SWATH-MS; antiviral immune response; clinical classifiers; high-throughput proteomics; mass spectrometry.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures

References
-
- Anderson N.L., Anderson N.G. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics. 2002;1:845–867. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
