

Quantitative Translation of Dog-to-Human Aging by Conserved Remodeling of the DNA Methylome
- PMID: 32619550
- PMCID: PMC7484147
- DOI: 10.1016/j.cels.2020.06.006
Quantitative Translation of Dog-to-Human Aging by Conserved Remodeling of the DNA Methylome
Abstract
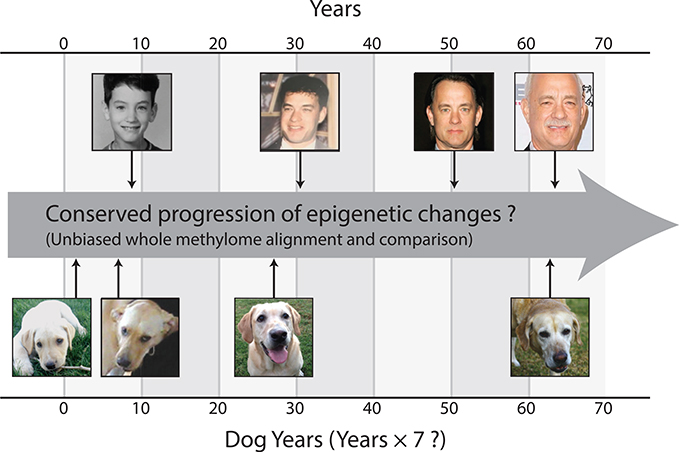
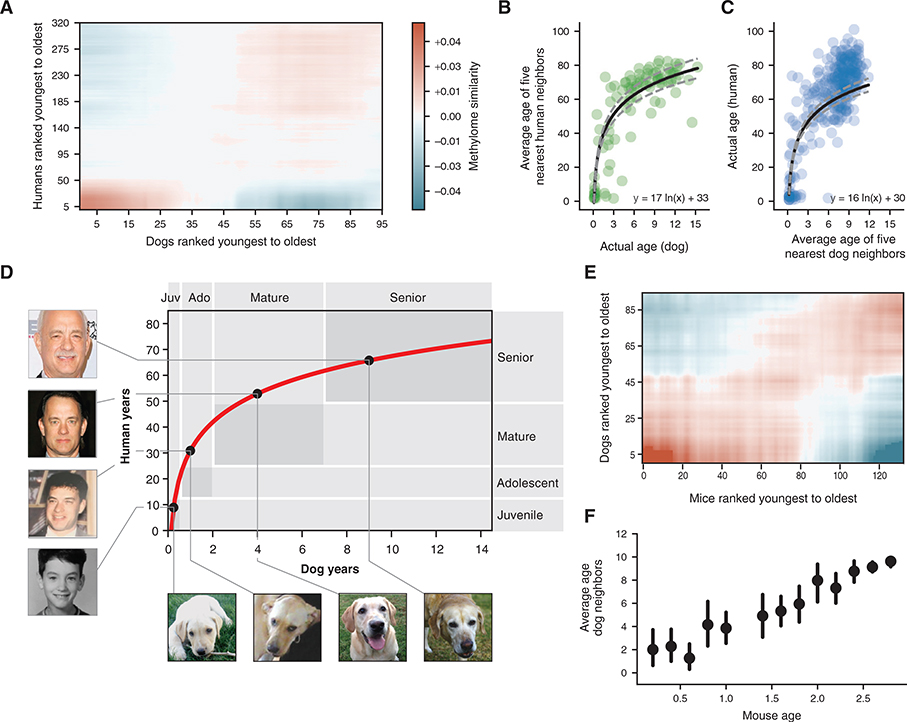
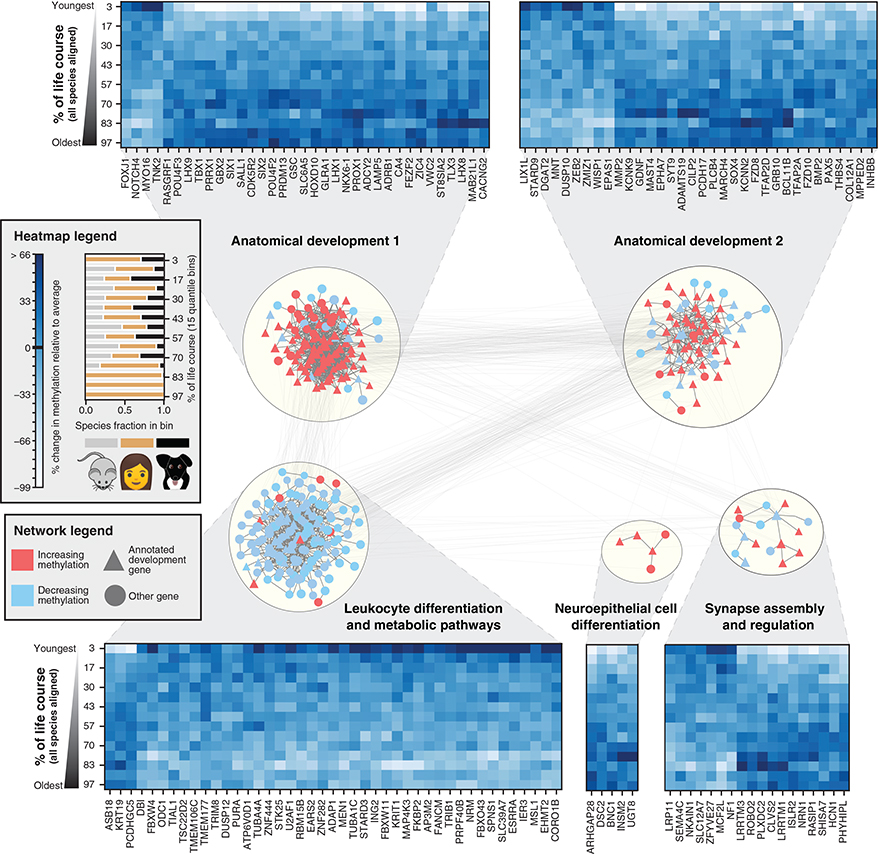
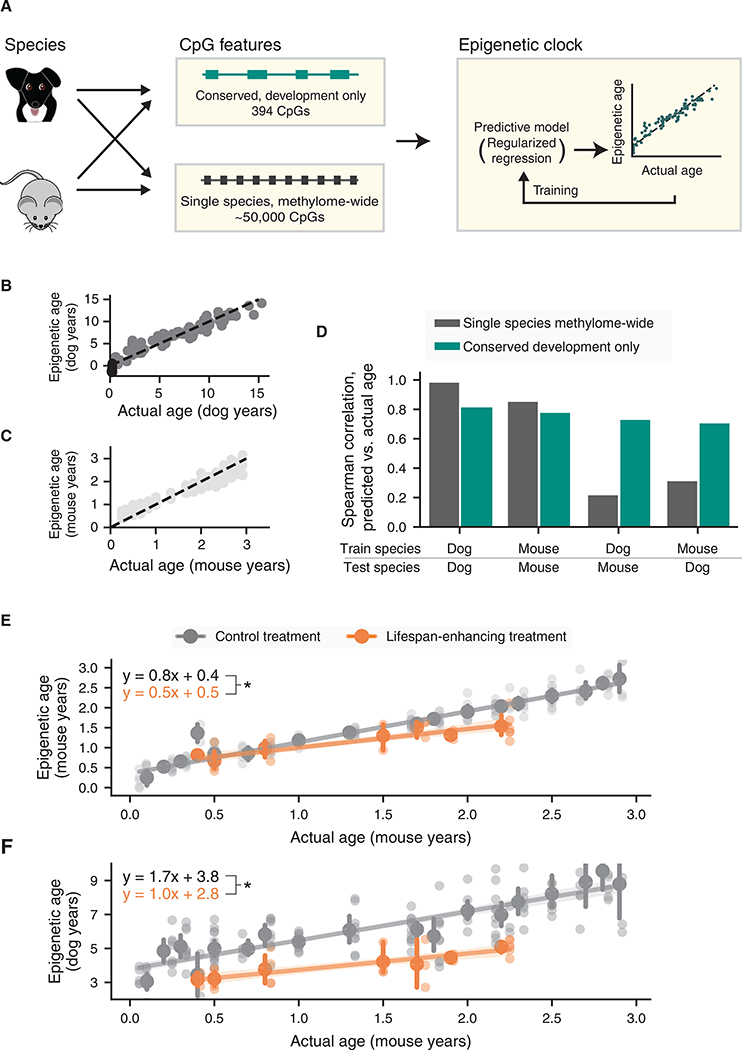
All mammals progress through similar physiological stages throughout life, from early development to puberty, aging, and death. Yet, the extent to which this conserved physiology reflects underlying genomic events is unclear. Here, we map the common methylation changes experienced by mammalian genomes as they age, focusing on comparison of humans with dogs, an emerging model of aging. Using oligo-capture sequencing, we characterize methylomes of 104 Labrador retrievers spanning a 16-year age range, achieving >150× coverage within mammalian syntenic blocks. Comparison with human methylomes reveals a nonlinear relationship that translates dog-to-human years and aligns the timing of major physiological milestones between the two species, with extension to mice. Conserved changes center on developmental gene networks, which are sufficient to translate age and the effects of anti-aging interventions across multiple mammals. These results establish methylation not only as a diagnostic age readout but also as a cross-species translator of physiological aging milestones.
Keywords: aging; canine; dog; epigenetic aging; epigenetic clock; epigenetics; epigenome; evolution; methylation; methylome.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests T.I. is co-founder of Data4Cure Inc, is on the Scientific Advisory Board, and has an equity interest. T.I. is on the Scientific Advisory Board of Ideaya Biosciences, has an equity interest, and receives sponsored research funding. The terms of these arrangements have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies. T.I. and T.W. hold a patent entitled “Methylome profiling of animals and uses thereof,” international patent application #PCT/US18/49103, US application #16/638,454.
Figures

Comment in
-
From dog days to human years.Nat Rev Genet. 2020 Sep;21(9):508-509. doi: 10.1038/s41576-020-0267-3. Nat Rev Genet. 2020. PMID: 32681033 No abstract available.
References
-
- Andrews S, et al. (2010). FastQC: a quality control tool for high throughput sequence data.
-
- Arias E, Heron M, and Xu J (2017). United States life tables, 2013. Natl Vital Stat. Rep. 66, 1–64. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
