

Enhancing droplet-based single-nucleus RNA-seq resolution using the semi-supervised machine learning classifier DIEM
- PMID: 32620816
- PMCID: PMC7335186
- DOI: 10.1038/s41598-020-67513-5
Enhancing droplet-based single-nucleus RNA-seq resolution using the semi-supervised machine learning classifier DIEM
Abstract
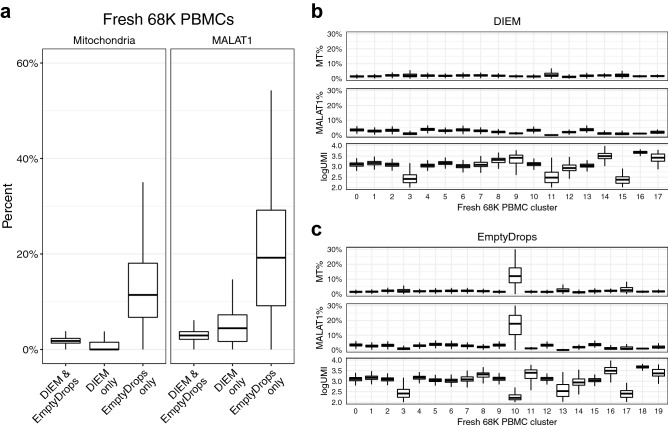
Single-nucleus RNA sequencing (snRNA-seq) measures gene expression in individual nuclei instead of cells, allowing for unbiased cell type characterization in solid tissues. We observe that snRNA-seq is commonly subject to contamination by high amounts of ambient RNA, which can lead to biased downstream analyses, such as identification of spurious cell types if overlooked. We present a novel approach to quantify contamination and filter droplets in snRNA-seq experiments, called Debris Identification using Expectation Maximization (DIEM). Our likelihood-based approach models the gene expression distribution of debris and cell types, which are estimated using EM. We evaluated DIEM using three snRNA-seq data sets: (1) human differentiating preadipocytes in vitro, (2) fresh mouse brain tissue, and (3) human frozen adipose tissue (AT) from six individuals. All three data sets showed evidence of extranuclear RNA contamination, and we observed that existing methods fail to account for contaminated droplets and led to spurious cell types. When compared to filtering using these state of the art methods, DIEM better removed droplets containing high levels of extranuclear RNA and led to higher quality clusters. Although DIEM was designed for snRNA-seq, our clustering strategy also successfully filtered single-cell RNA-seq data. To conclude, our novel method DIEM removes debris-contaminated droplets from single-cell-based data fast and effectively, leading to cleaner downstream analysis. Our code is freely available for use at https://github.com/marcalva/diem.
Conflict of interest statement
The authors declare no competing interests.
Figures
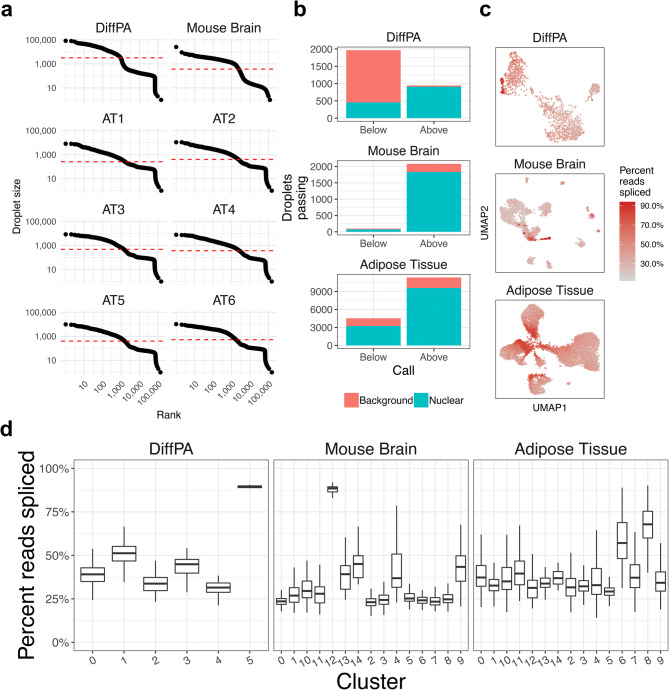
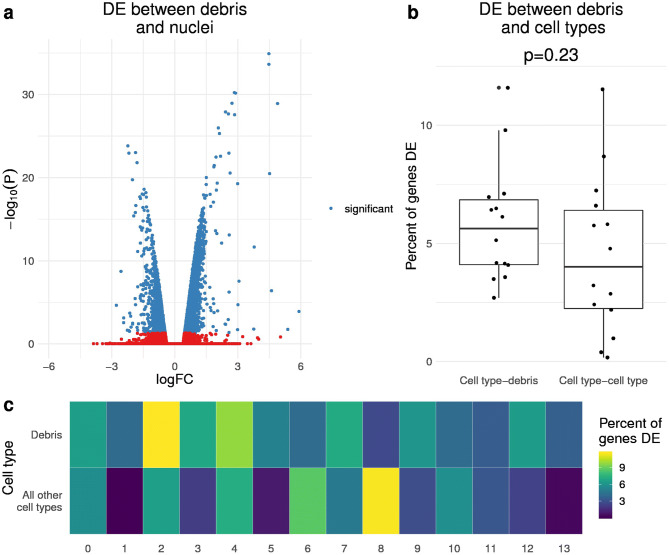

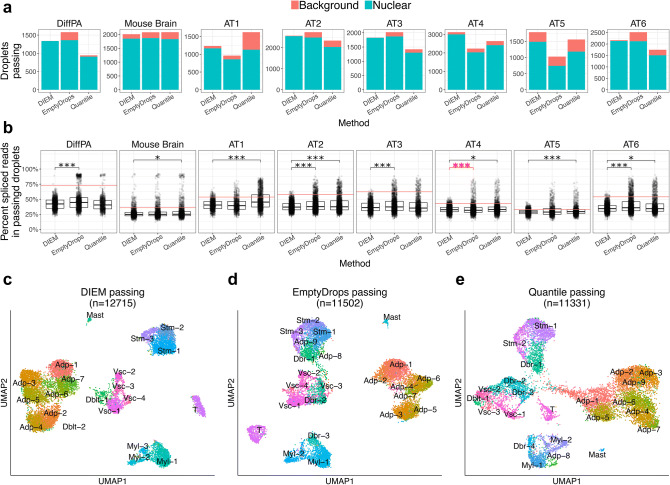
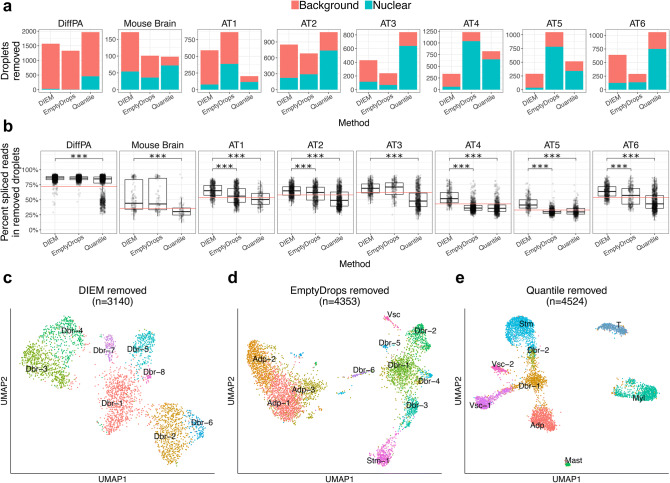
References
Publication types
MeSH terms
Grants and funding
- R25 GM112625/GM/NIGMS NIH HHS/United States
- R01 MH122569/MH/NIMH NIH HHS/United States
- R01 HG010505/HG/NHGRI NIH HHS/United States
- P01 HL028481/HL/NHLBI NIH HHS/United States
- P30 DK041301/DK/NIDDK NIH HHS/United States
- F31 HL142180/HL/NHLBI NIH HHS/United States
- UL1 TR001881/TR/NCATS NIH HHS/United States
- R56 MD013312/MD/NIMHD NIH HHS/United States
- R01 MH115979/MH/NIMH NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- T32 HG002536/HG/NHGRI NIH HHS/United States
- U01 DK105561/DK/NIDDK NIH HHS/United States
- R01 HL095056/HL/NHLBI NIH HHS/United States
