

Osteogenesis imperfecta: an update on clinical features and therapies
- PMID: 32621590
- PMCID: PMC7694877
- DOI: 10.1530/EJE-20-0299
Osteogenesis imperfecta: an update on clinical features and therapies
Abstract
Osteogenesis imperfecta (OI) is an inherited skeletal dysplasia characterized by bone fragility and skeletal deformities. While the majority of cases are associated with pathogenic variants in COL1A1 and COL1A2, the genes encoding type I collagen, up to 25% of cases are associated with other genes that function within the collagen biosynthesis pathway or are involved in osteoblast differentiation and bone mineralization. Clinically, OI is heterogeneous in features and variable in severity. In addition to the skeletal findings, it can affect multiple systems including dental and craniofacial abnormalities, muscle weakness, hearing loss, respiratory and cardiovascular complications. A multi-disciplinary approach to care is recommended to address not only the fractures, reduced mobility, growth and bone pain but also other extra-skeletal manifestations. While bisphosphonates remain the mainstay of treatment in OI, new strategies are being explored, such as sclerostin inhibitory antibodies and TGF beta inhibition, to address not only the low bone mineral density but also the inherent bone fragility. Studies in animal models have expanded the understanding of pathomechanisms of OI and, along with ongoing clinical trials, will allow to develop better therapeutic approaches for these patients.
Conflict of interest statement
Declaration of interest
The authors declare no conflicts of interest.
Figures
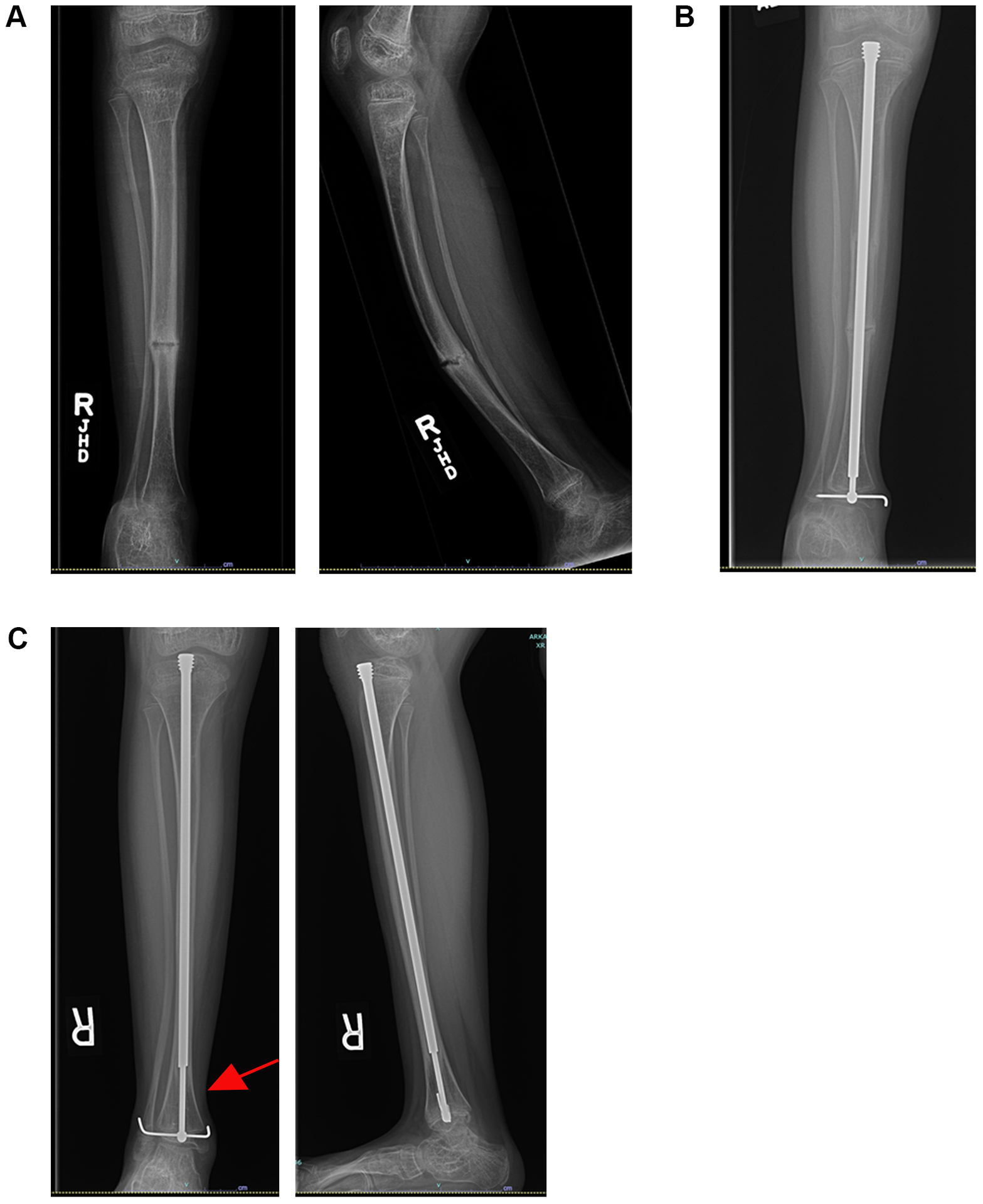
References
-
- Marini JC, Forlino A, Bachinger HP, Bishop NJ, Byers PH, Paepe A, Fassier F, Fratzl-Zelman N, Kozloff KM, Krakow D et al. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017. Aug 18;3:17052. - PubMed
-
- Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, Nishimura G, Robertson S, Sangiorgi L, Savarirayan R et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019. Dec;179(12):2393–419. - PubMed
-
- Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J, Iden S, Wirth B, Eysel P, Koerber F et al. A mutation in the 5’-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet. 2012. August 10;91(2):349–57. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
