

Photo-SNAP-tag, a Light-Regulated Chemical Labeling System
- PMID: 32623878
- PMCID: PMC7596905
- DOI: 10.1021/acschembio.0c00412
Photo-SNAP-tag, a Light-Regulated Chemical Labeling System
Abstract
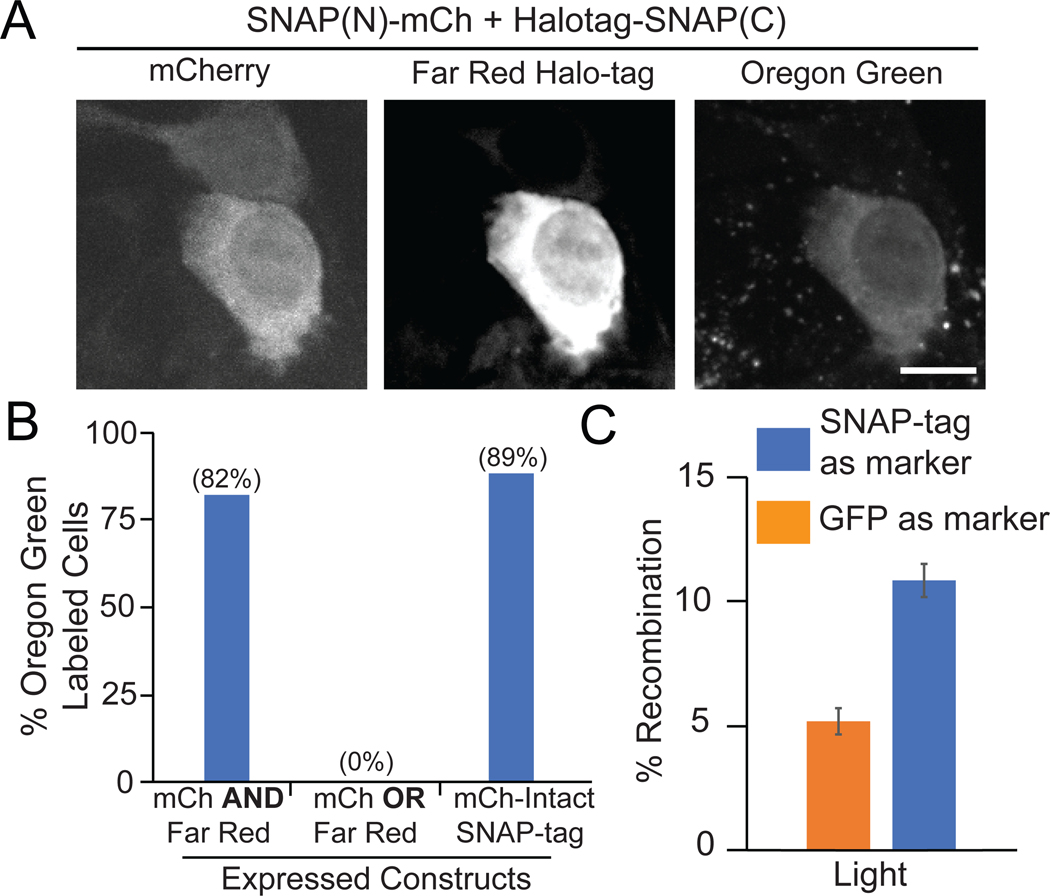
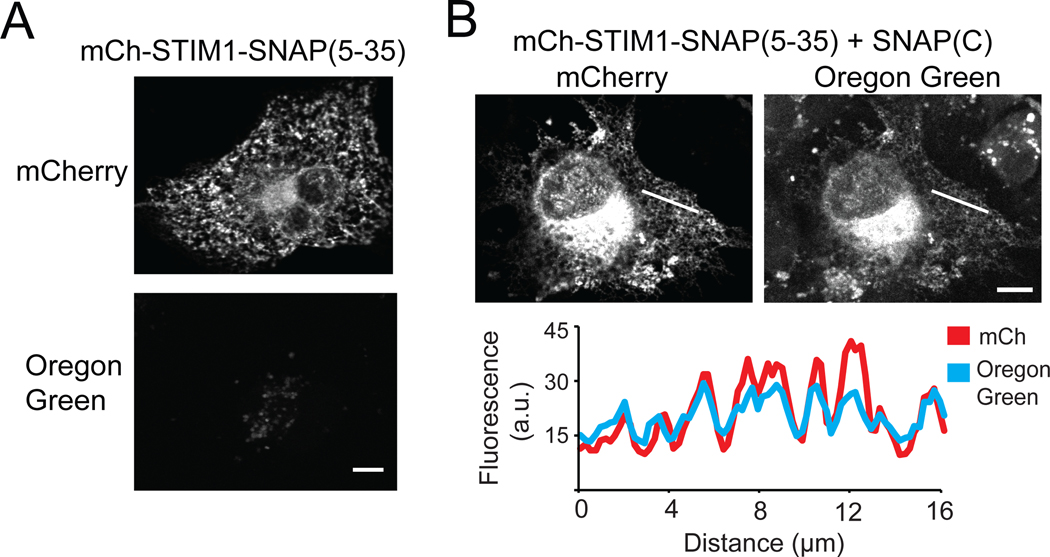
Methods that allow labeling and tracking of proteins have been instrumental for understanding their function. Traditional methods for labeling proteins include fusion to fluorescent proteins or self-labeling chemical tagging systems such as SNAP-tag or Halo-tag. These latter approaches allow bright fluorophores or other chemical moieties to be attached to a protein of interest through a small fusion tag. In this work, we sought to improve the versatility of self-labeling chemical-tagging systems by regulating their activity with light. We used light-inducible dimerizers to reconstitute a split SNAP-tag (modified human O6-alkylguanine-DNA-alkyltransferase, hAGT) protein, allowing tight light-dependent control of chemical labeling. In addition, we generated a small split SNAP-tag fragment that can efficiently self-assemble with its complement fragment, allowing high labeling efficacy with a small tag. We envision these tools will extend the versatility and utility of the SNAP-tag chemical system for protein labeling applications.
Figures
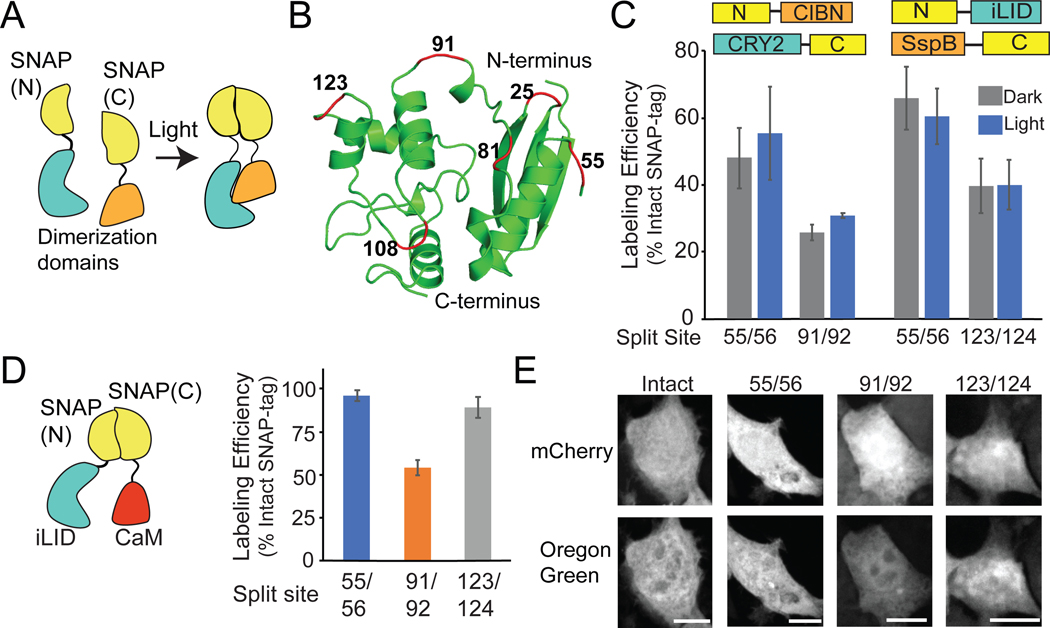
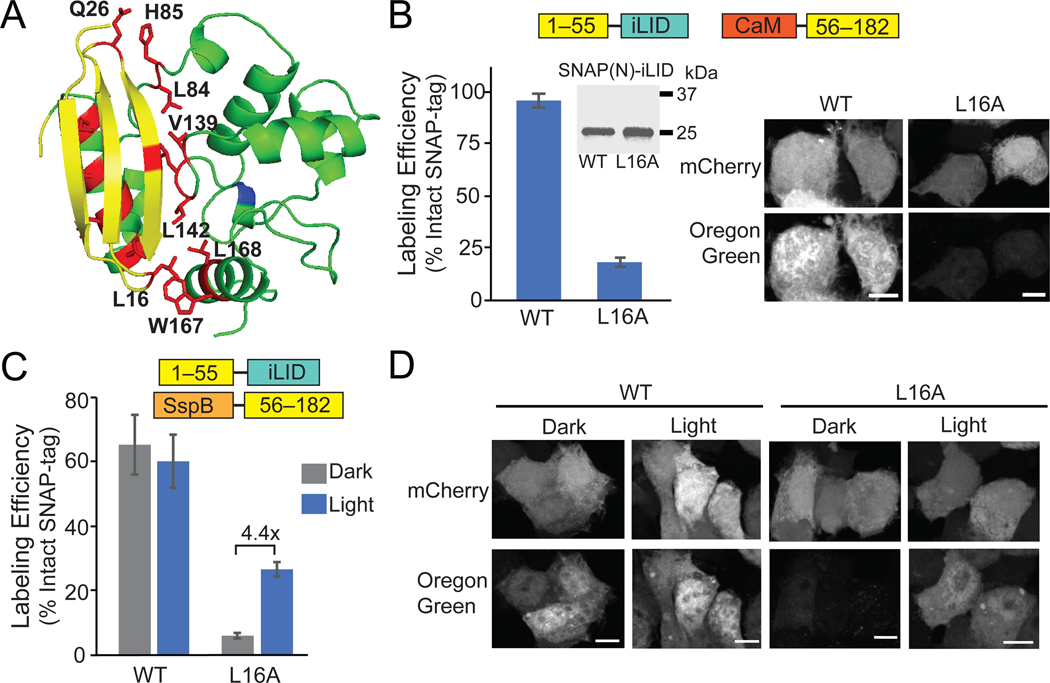
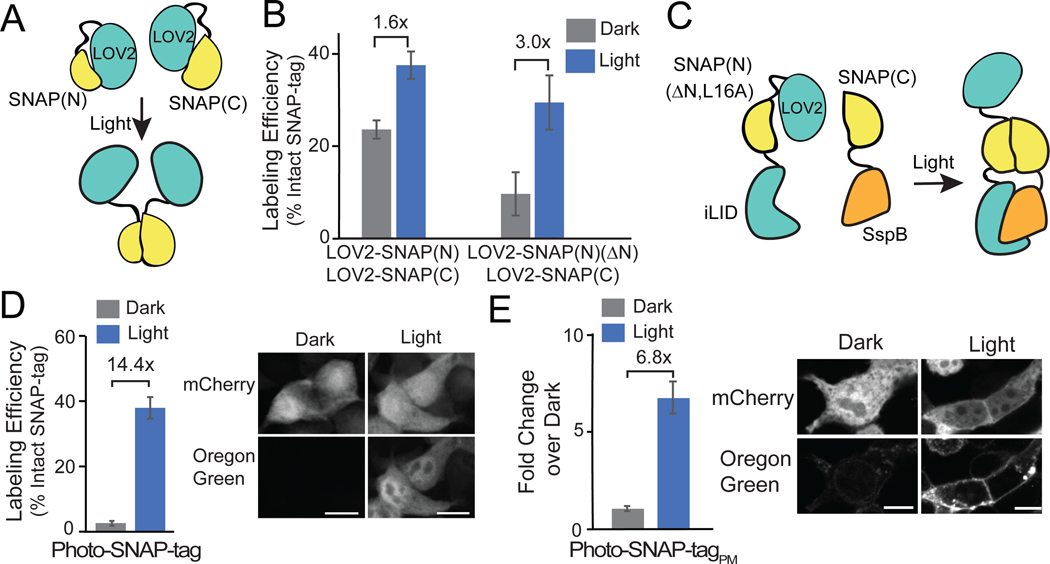
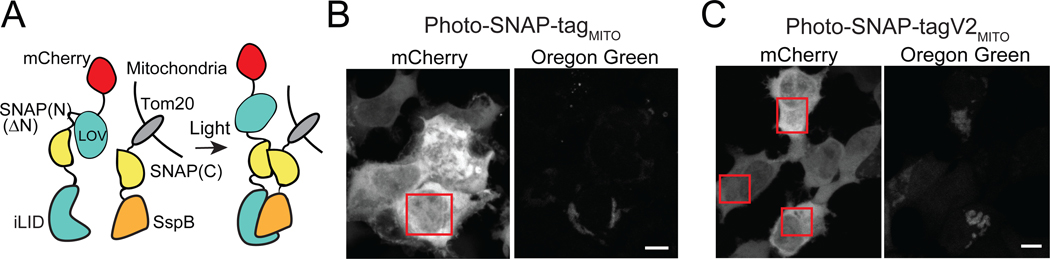
References
-
- Patterson GH, and Lippincott-Schwartz J. (2002) A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297, 1873–1877. - PubMed
-
- Adam V, Berardozzi R, Byrdin M, and Bourgeois D. (2014) Phototransformable fluorescent proteins: Future challenges. Curr. Opin. Chem. Biol. 20, 92–102. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
