

Exciting Complexity: The Role of Motor Circuit Elements in ALS Pathophysiology
- PMID: 32625051
- PMCID: PMC7311855
- DOI: 10.3389/fnins.2020.00573
Exciting Complexity: The Role of Motor Circuit Elements in ALS Pathophysiology
Abstract
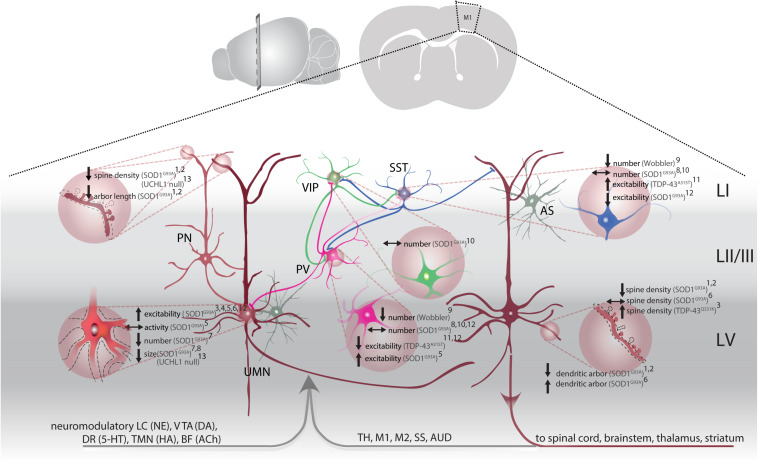
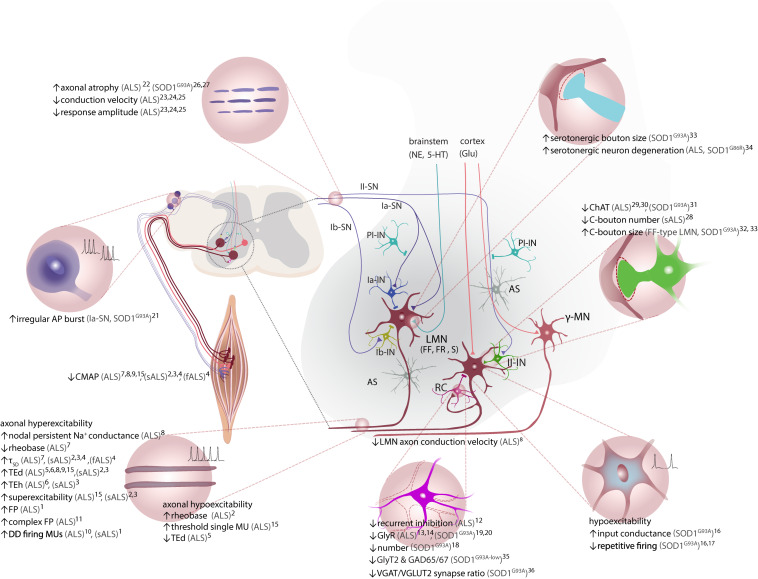
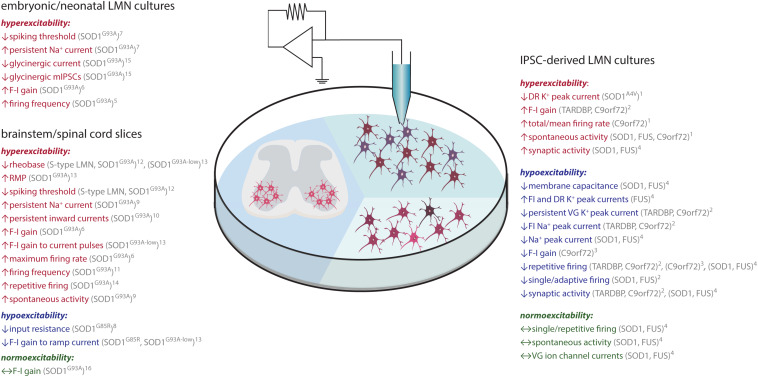
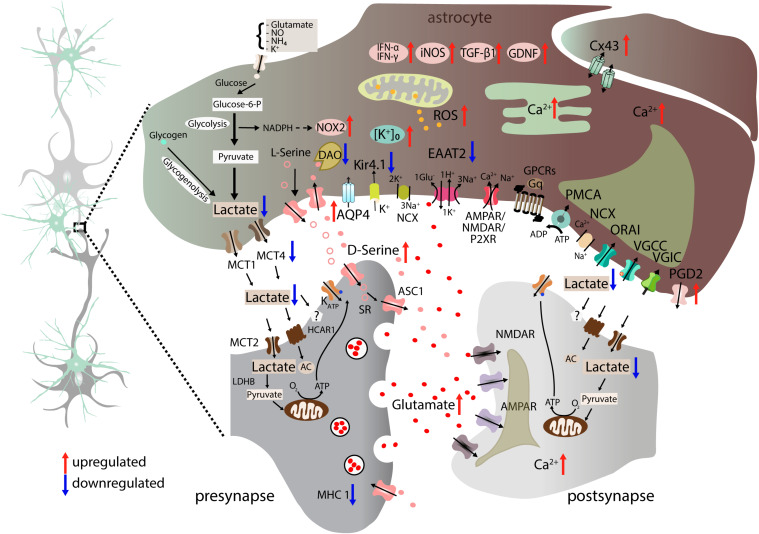
Amyotrophic lateral sclerosis (ALS) is a fatal disease, characterized by the degeneration of both upper and lower motor neurons. Despite decades of research, we still to date lack a cure or disease modifying treatment, emphasizing the need for a much-improved insight into disease mechanisms and cell type vulnerability. Altered neuronal excitability is a common phenomenon reported in ALS patients, as well as in animal models of the disease, but the cellular and circuit processes involved, as well as the causal relevance of those observations to molecular alterations and final cell death, remain poorly understood. Here, we review evidence from clinical studies, cell type-specific electrophysiology, genetic manipulations and molecular characterizations in animal models and culture experiments, which argue for a causal involvement of complex alterations of structure, function and connectivity of different neuronal subtypes within the cortical and spinal cord motor circuitries. We also summarize the current knowledge regarding the detrimental role of astrocytes and reassess the frequently proposed hypothesis of glutamate-mediated excitotoxicity with respect to changes in neuronal excitability. Together, these findings suggest multifaceted cell type-, brain area- and disease stage- specific disturbances of the excitation/inhibition balance as a cardinal aspect of ALS pathophysiology.
Keywords: Amyotrophic lateral sclerosis; astrocytes; excitability; excitotoxicity; interneurons; lower motor neurons; neural circuits; upper motor neurons.
Copyright © 2020 Gunes, Kan, Ye and Liebscher.
Figures
References
-
- Abe K., Itoyama Y., Sobue G., Tsuji S., Aoki M., Doyu M., et al. (2014). Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Frontotemporal. Degener. 15 610–617. 10.3109/21678421.2014.959024 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous