

A network-based integrated framework for predicting virus-prokaryote interactions
- PMID: 32626849
- PMCID: PMC7324143
- DOI: 10.1093/nargab/lqaa044
A network-based integrated framework for predicting virus-prokaryote interactions
Abstract
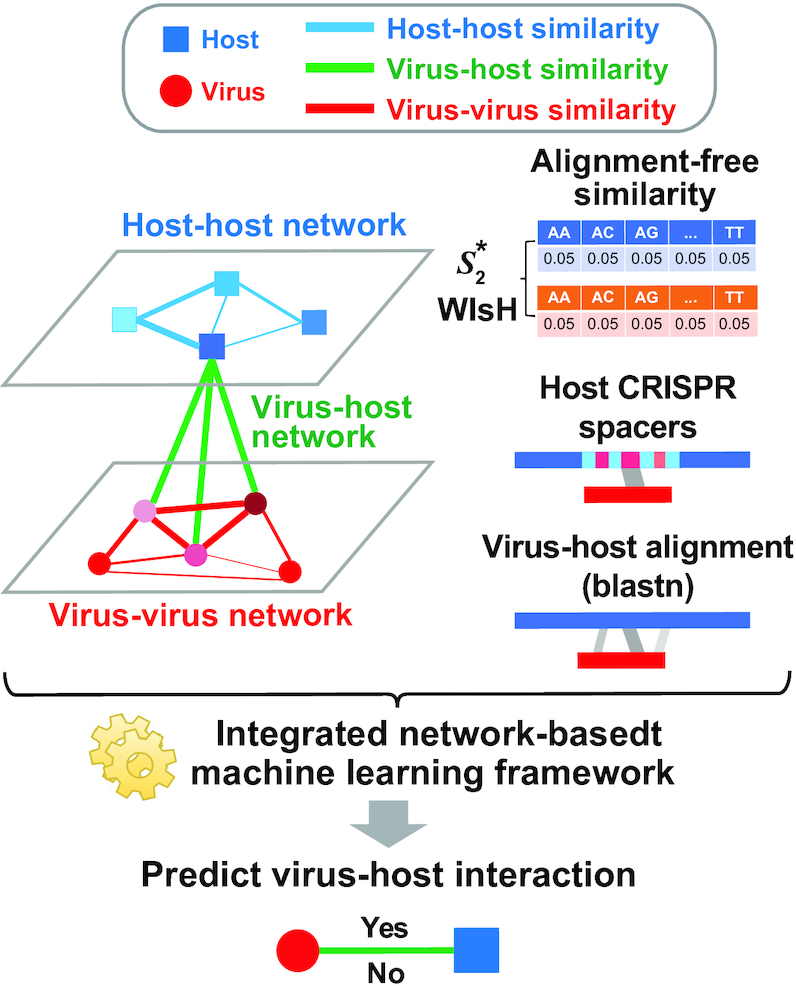
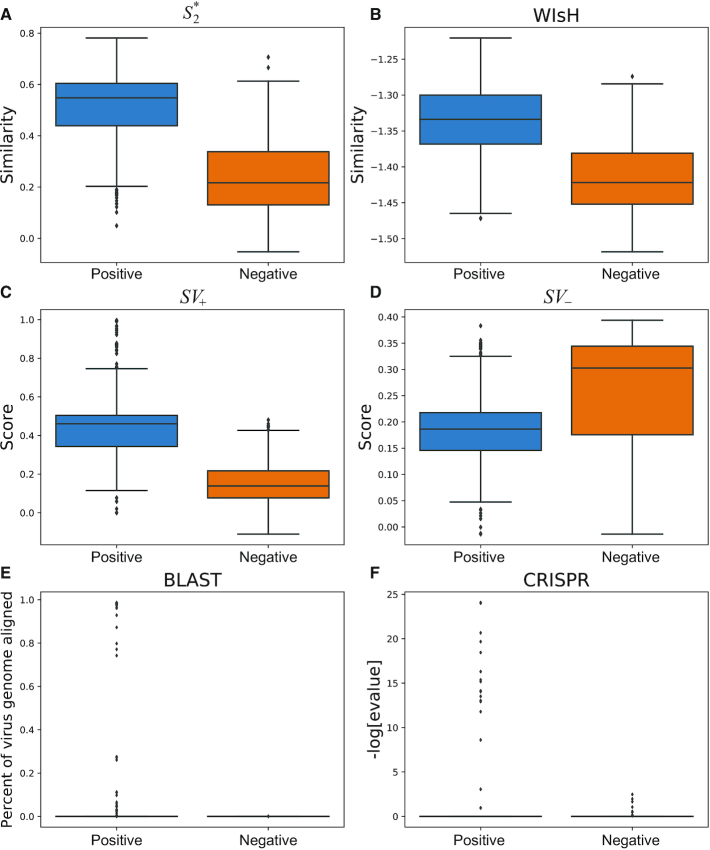
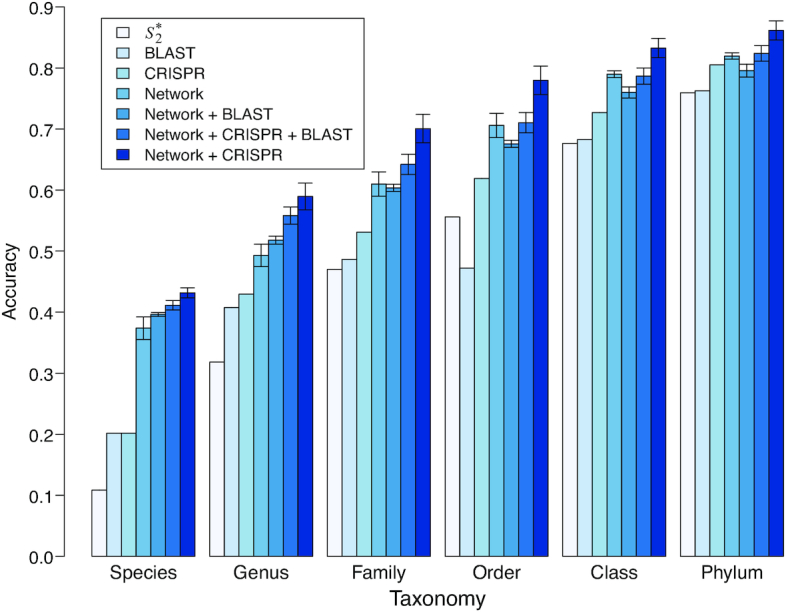
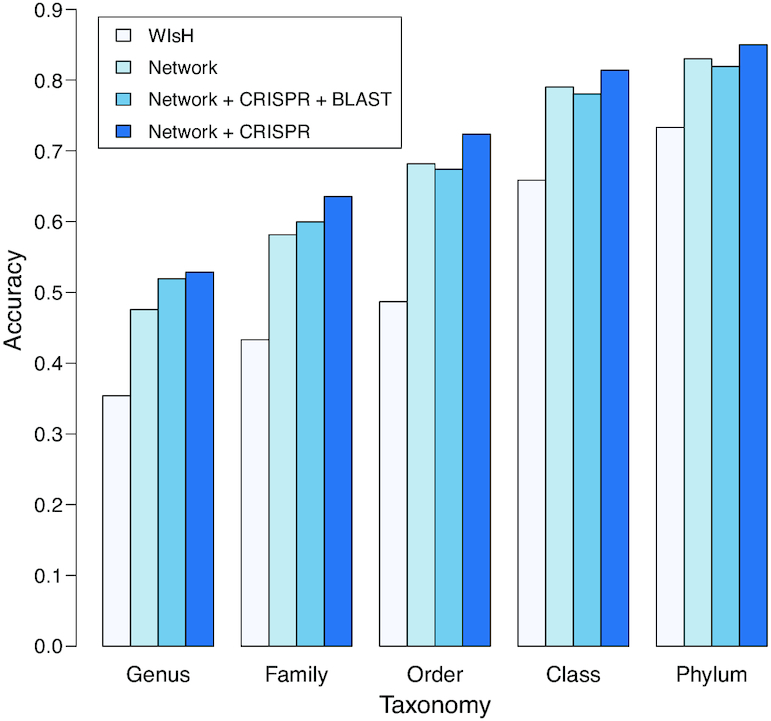
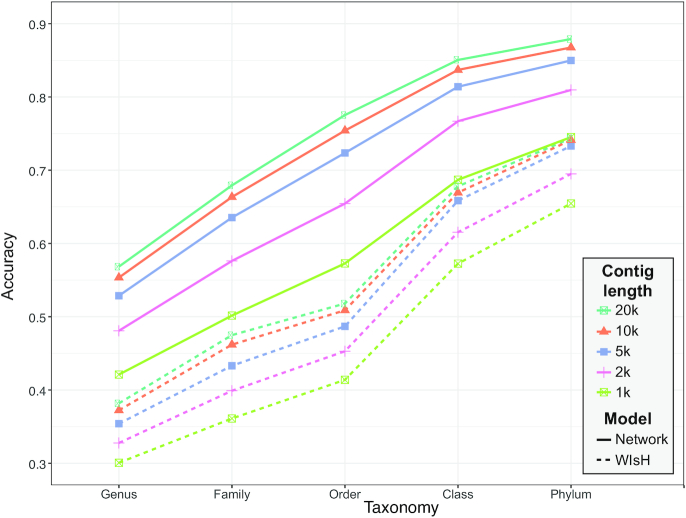
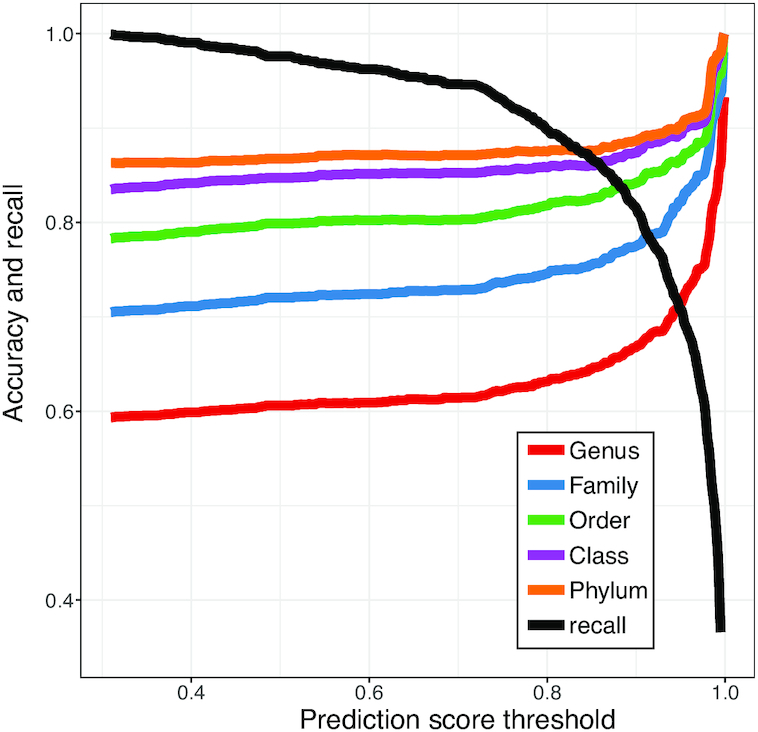
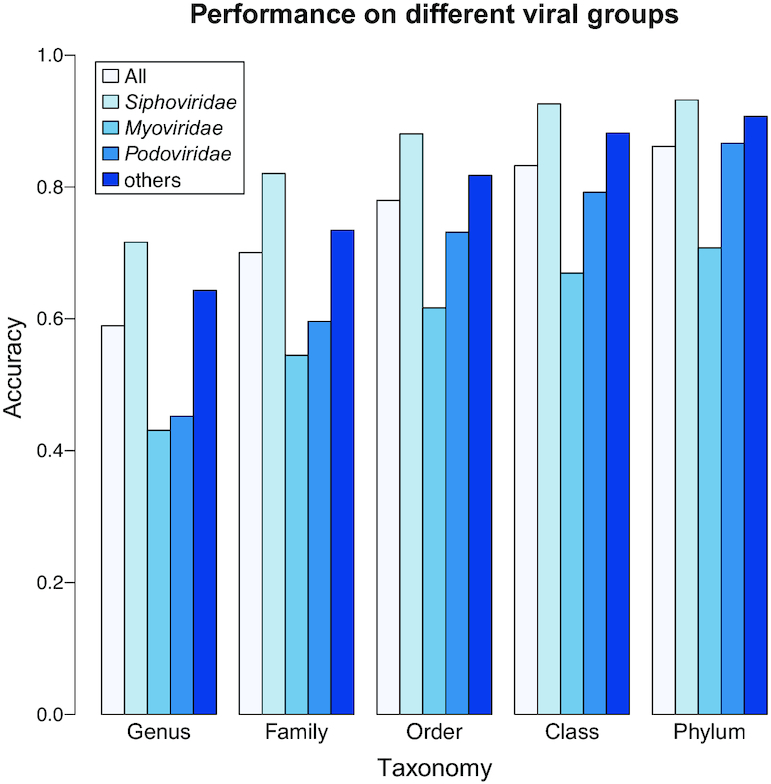
Metagenomic sequencing has greatly enhanced the discovery of viral genomic sequences; however, it remains challenging to identify the host(s) of these new viruses. We developed VirHostMatcher-Net, a flexible, network-based, Markov random field framework for predicting virus-prokaryote interactions using multiple, integrated features: CRISPR sequences and alignment-free similarity measures ([Formula: see text] and WIsH). Evaluation of this method on a benchmark set of 1462 known virus-prokaryote pairs yielded host prediction accuracy of 59% and 86% at the genus and phylum levels, representing 16-27% and 6-10% improvement, respectively, over previous single-feature prediction approaches. We applied our host prediction tool to crAssphage, a human gut phage, and two metagenomic virus datasets: marine viruses and viral contigs recovered from globally distributed, diverse habitats. Host predictions were frequently consistent with those of previous studies, but more importantly, this new tool made many more confident predictions than previous tools, up to nearly 3-fold more (n > 27 000), greatly expanding the diversity of known virus-host interactions.
© The Author(s) 2019. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures
References
-
- Breitbart M., Rohwer F.. Here a virus, there a virus, everywhere the same virus?. Trends Microbiol. 2005; 13:278–284. - PubMed
-
- Fierer N., Breitbart M., Nulton J., Salamon P., Lozupone C., Jones R., Robeson M., Edwards R.A., Felts B., Rayhawk S. et al. .. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl. Environ. Microb. 2007; 73:7059–7066. - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
