

ZipSeq: barcoding for real-time mapping of single cell transcriptomes
- PMID: 32632238
- PMCID: PMC7891292
- DOI: 10.1038/s41592-020-0880-2
ZipSeq: barcoding for real-time mapping of single cell transcriptomes
Abstract
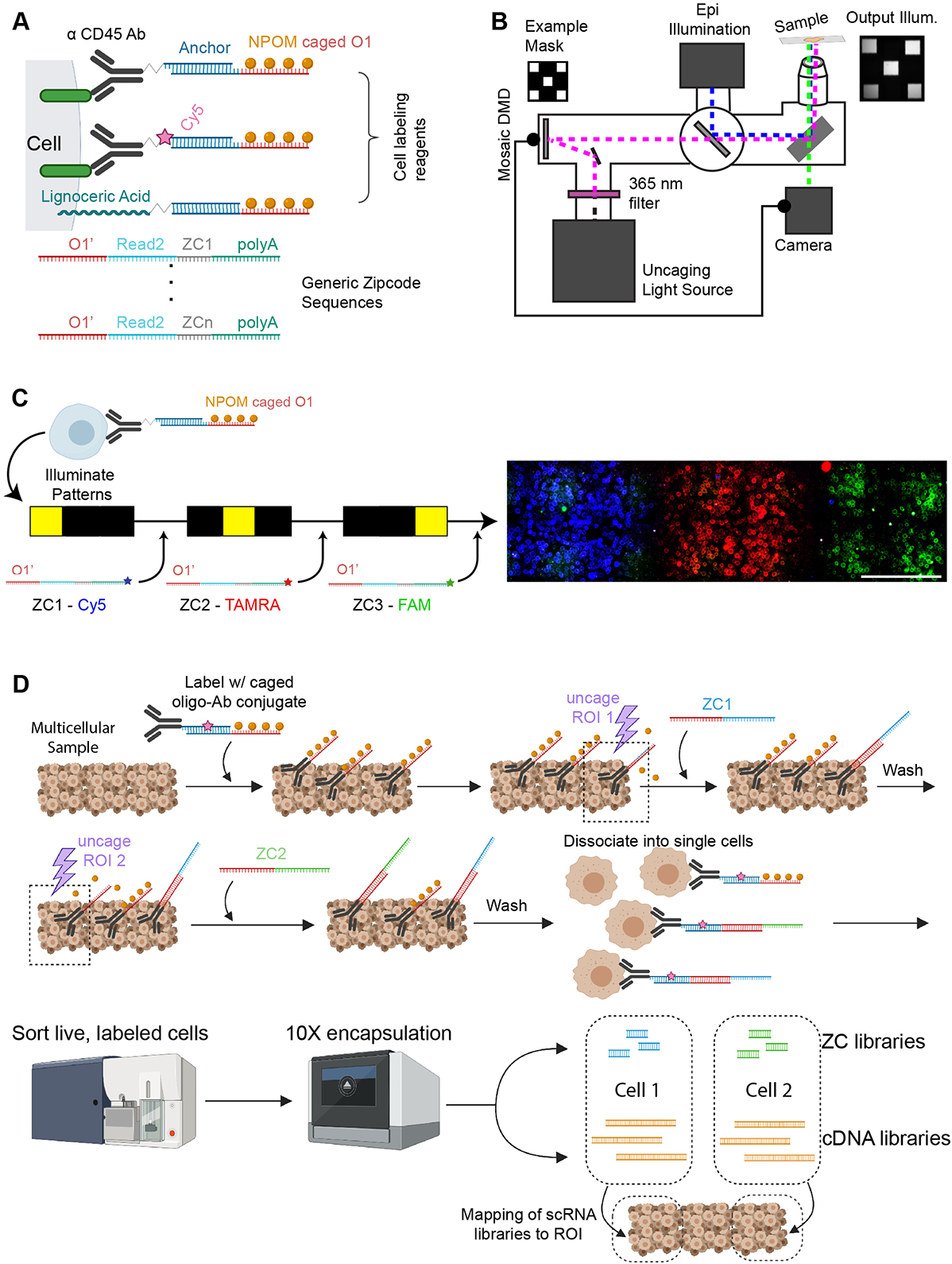
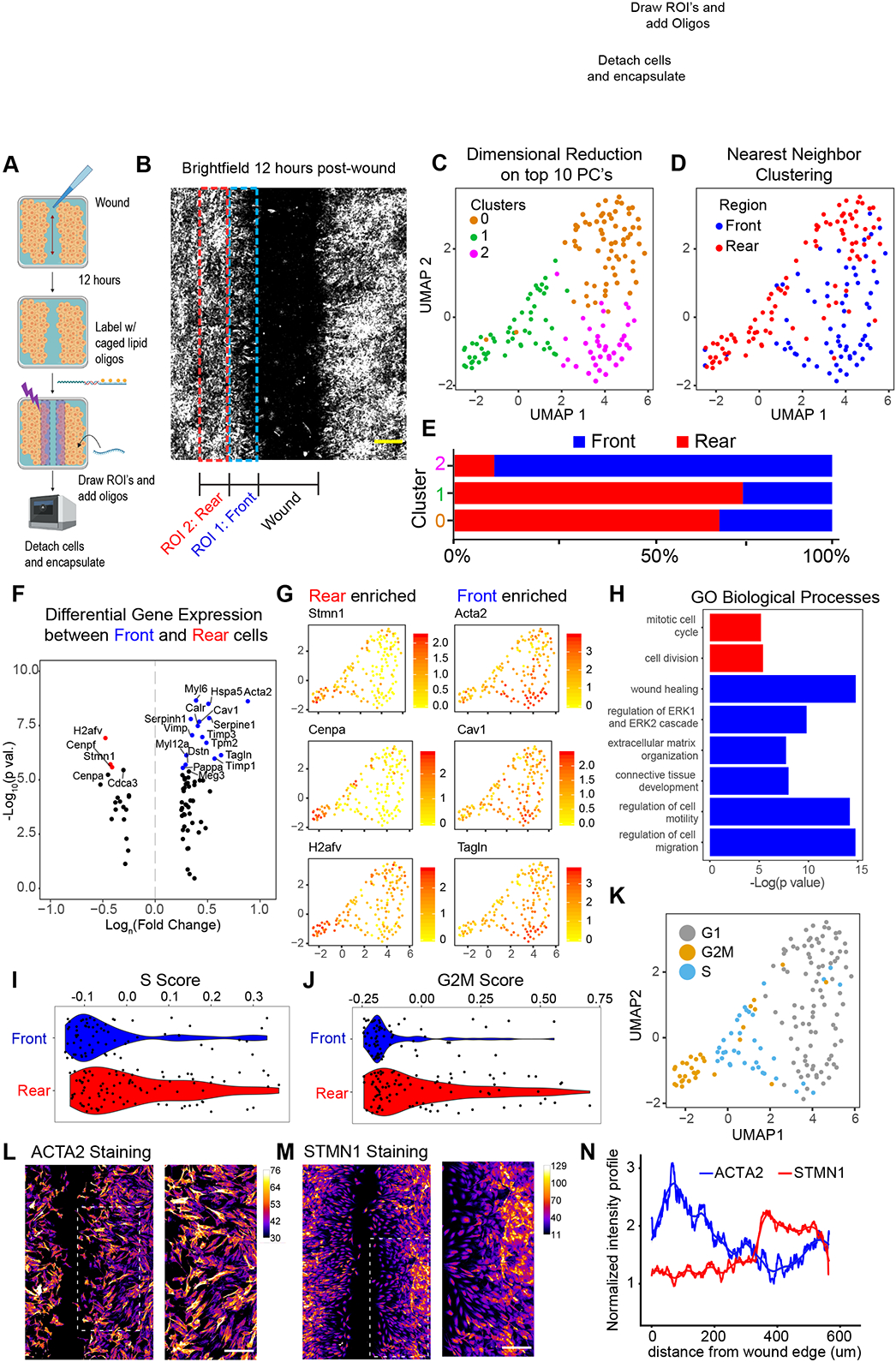
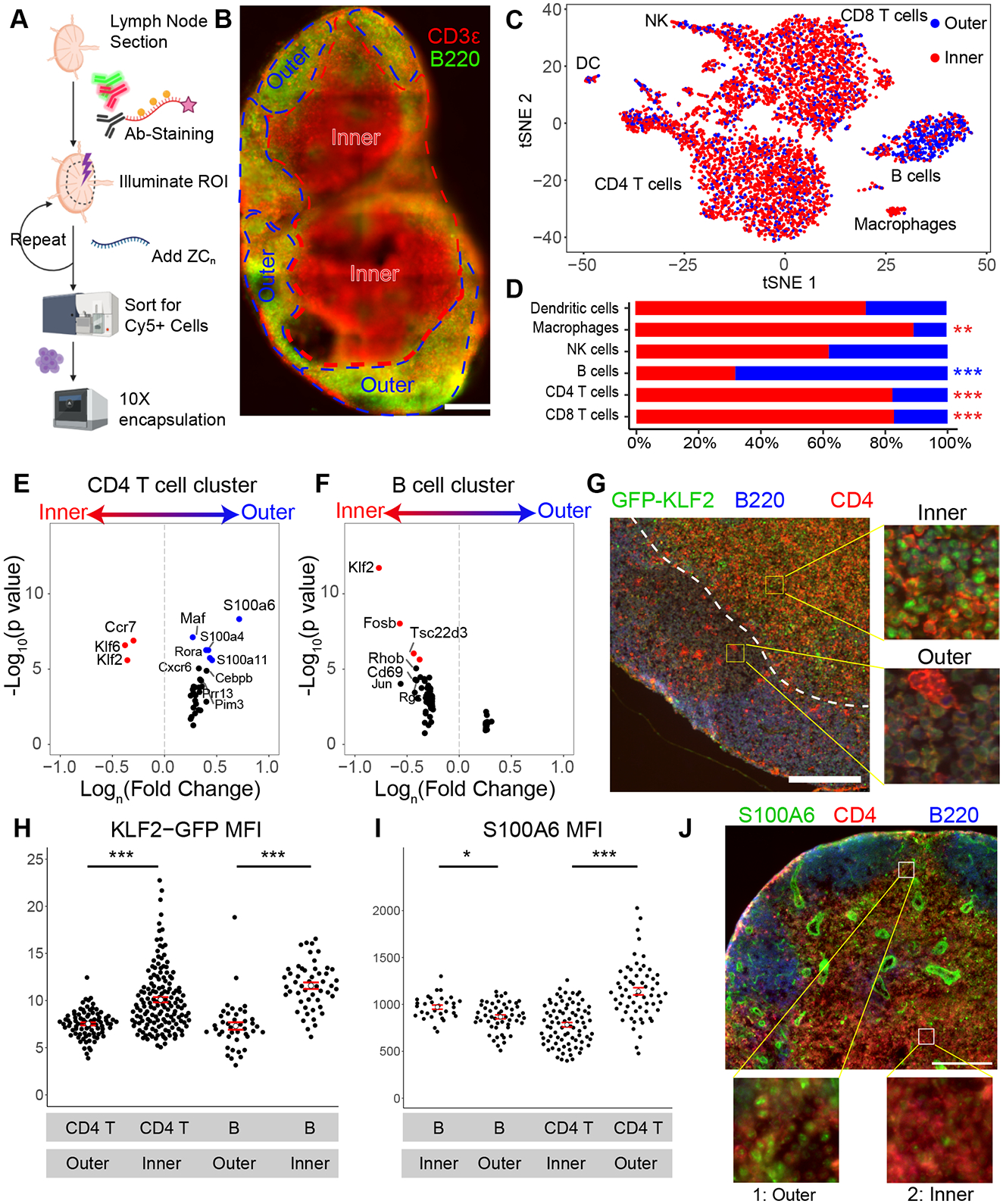
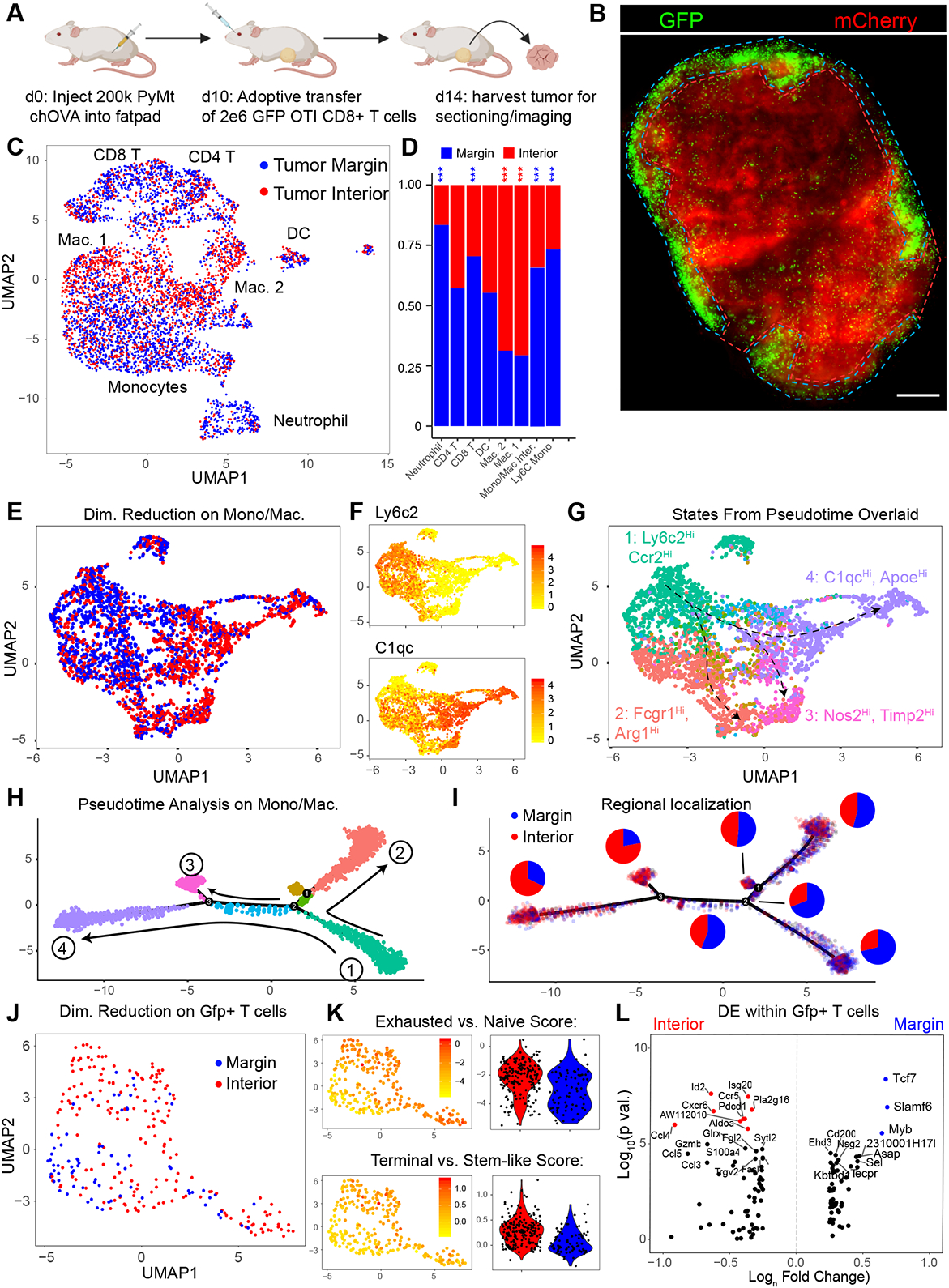
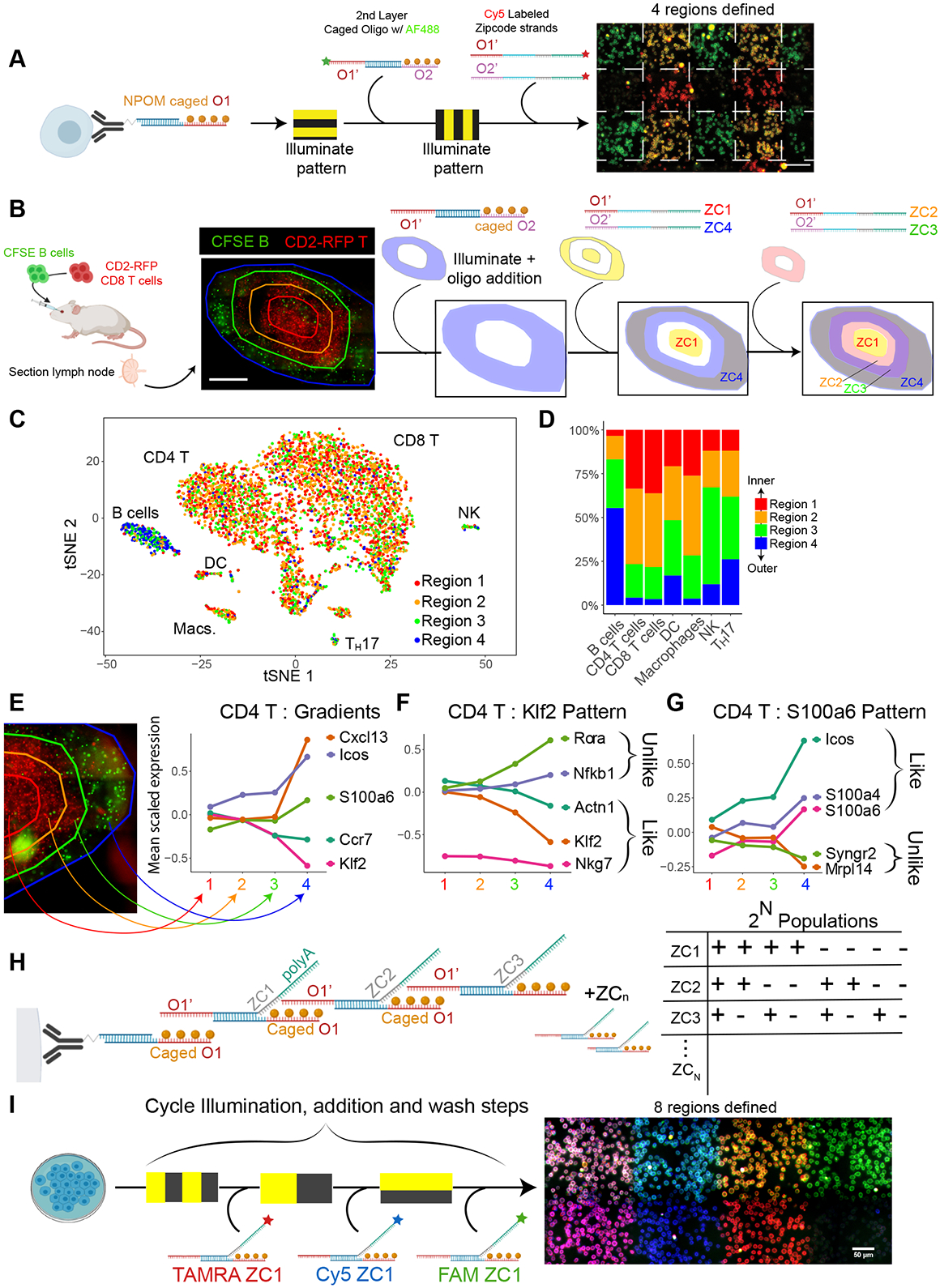
Spatial transcriptomics seeks to integrate single cell transcriptomic data within the three-dimensional space of multicellular biology. Current methods to correlate a cell's position with its transcriptome in living tissues have various limitations. We developed an approach, called 'ZipSeq', that uses patterned illumination and photocaged oligonucleotides to serially print barcodes ('zipcodes') onto live cells in intact tissues, in real time and with an on-the-fly selection of patterns. Using ZipSeq, we mapped gene expression in three settings: in vitro wound healing, live lymph node sections and a live tumor microenvironment. In all cases, we discovered new gene expression patterns associated with histological structures. In the tumor microenvironment, this demonstrated a trajectory of myeloid and T cell differentiation from the periphery inward. A combinatorial variation of ZipSeq efficiently scales in the number of regions defined, providing a pathway for complete mapping of live tissues, subsequent to real-time imaging or perturbation.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS
K.H.H. and M.F.K. are listed on a patent application regarding the ZipSeq approach.
Figures
Comment in
-
A zipcode for transcriptomes.Nat Rev Genet. 2020 Sep;21(9):508-509. doi: 10.1038/s41576-020-0266-4. Nat Rev Genet. 2020. PMID: 32661360 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
