

RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells
- PMID: 32632336
- PMCID: PMC10353856
- DOI: 10.1038/s41588-020-0662-x
RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells
Abstract
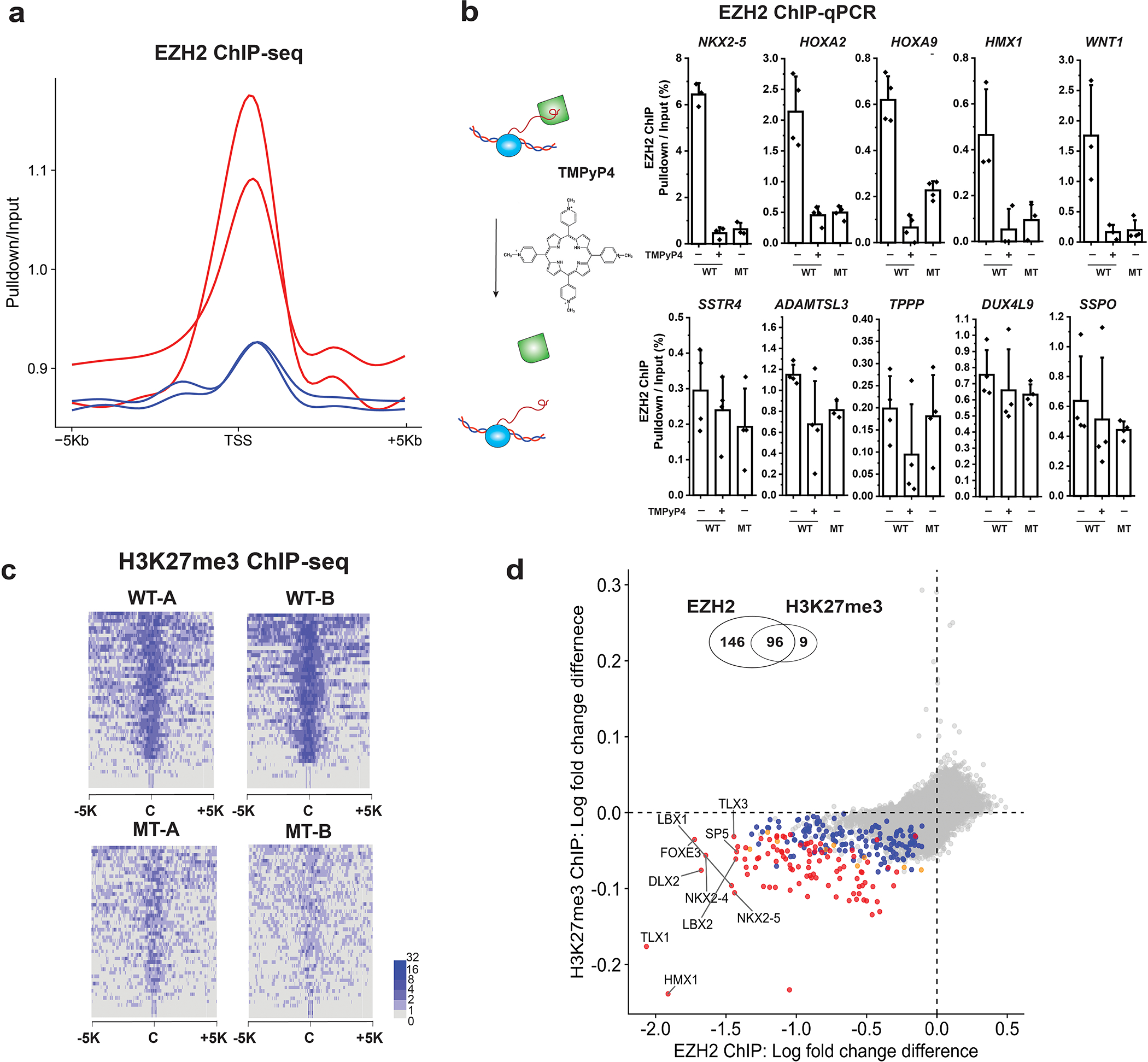
Many chromatin-binding proteins and protein complexes that regulate transcription also bind RNA. One of these, Polycomb repressive complex 2 (PRC2), deposits the H3K27me3 mark of facultative heterochromatin and is required for stem cell differentiation. PRC2 binds RNAs broadly in vivo and in vitro. Yet, the biological importance of this RNA binding remains unsettled. Here, we tackle this question in human induced pluripotent stem cells by using multiple complementary approaches. Perturbation of RNA-PRC2 interaction by RNase A, by a chemical inhibitor of transcription or by an RNA-binding-defective mutant all disrupted PRC2 chromatin occupancy and localization genome wide. The physiological relevance of PRC2-RNA interactions is further underscored by a cardiomyocyte differentiation defect upon genetic disruption. We conclude that PRC2 requires RNA binding for chromatin localization in human pluripotent stem cells and in turn for defining cellular state.
Conflict of interest statement
Competing interests
T.R.C. is on the Merck board and is a consultant for Storm Therapeutics.
Figures













Comment in
-
RNA closing the Polycomb circle.Nat Genet. 2020 Sep;52(9):866-867. doi: 10.1038/s41588-020-0683-5. Nat Genet. 2020. PMID: 32879510 No abstract available.
References
-
- Cao R et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043 (2002). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
