

MEK activation modulates glycolysis and supports suppressive myeloid cells in TNBC
- PMID: 32634121
- PMCID: PMC7455066
- DOI: 10.1172/jci.insight.134290
MEK activation modulates glycolysis and supports suppressive myeloid cells in TNBC
Abstract
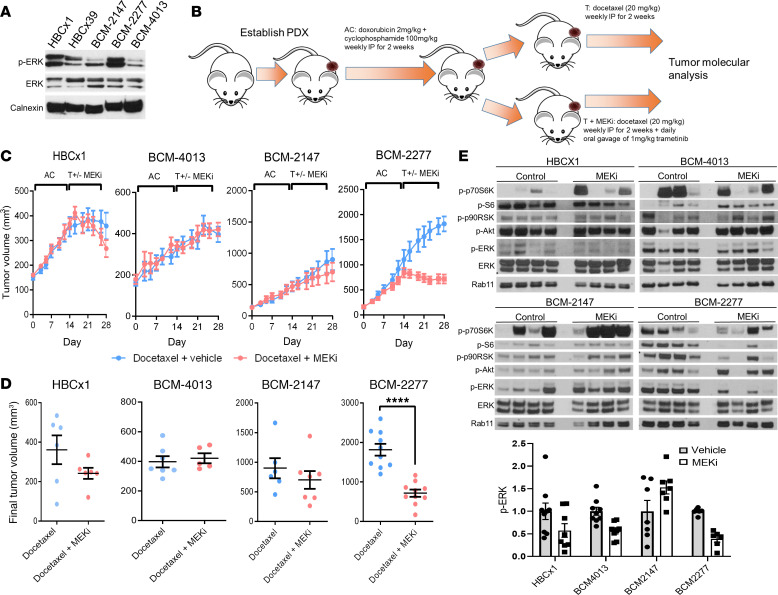
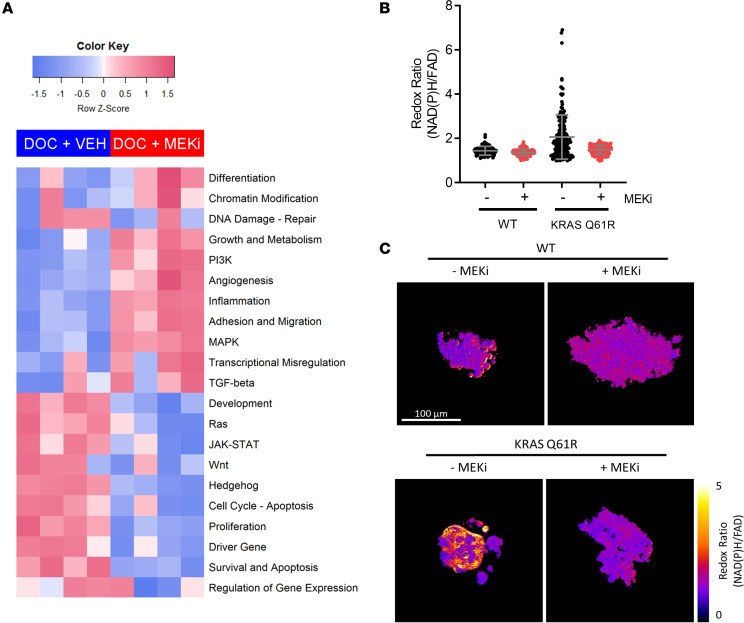
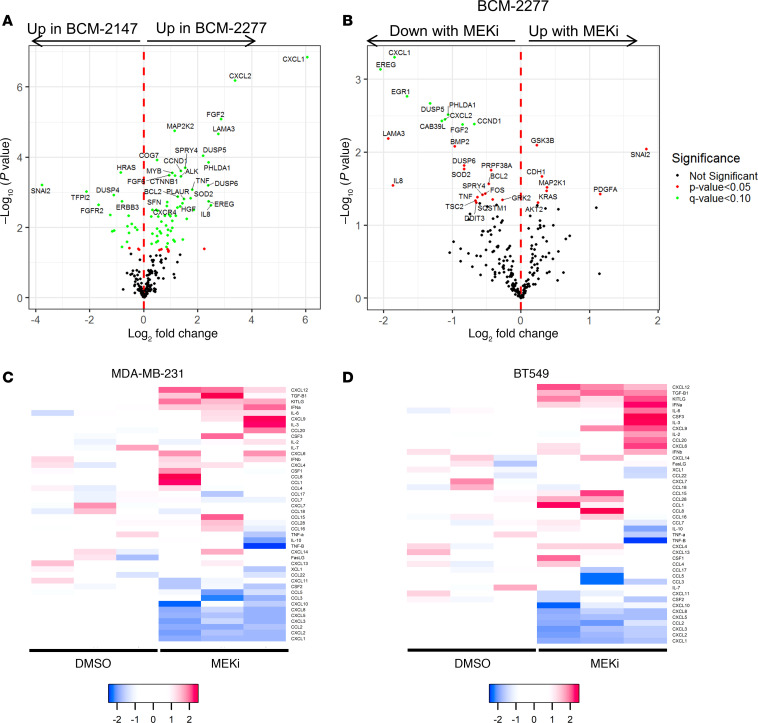
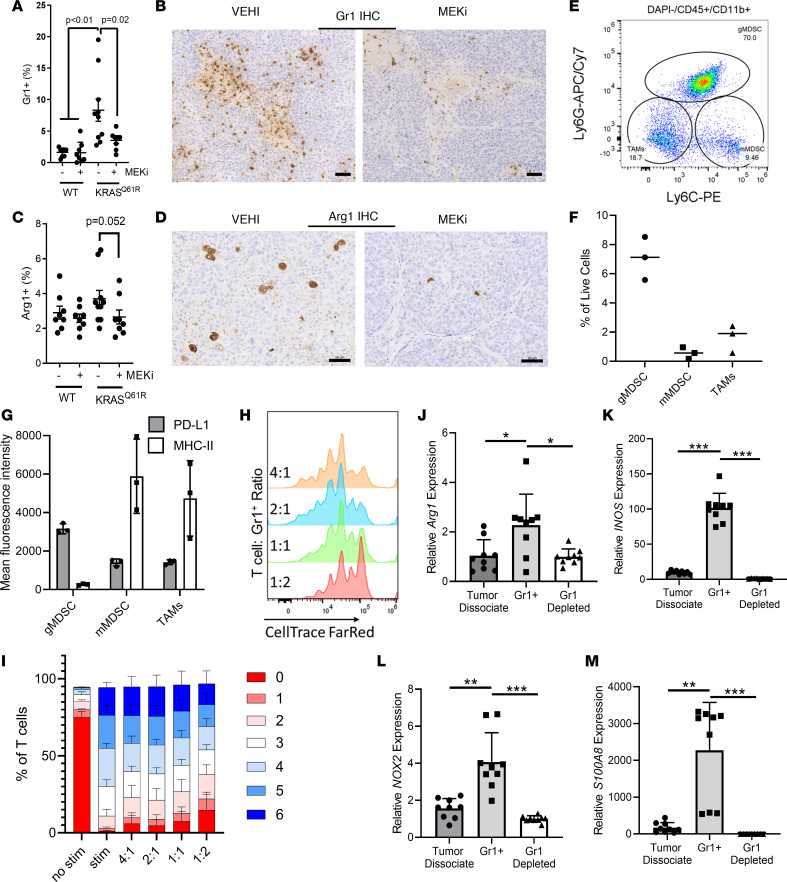
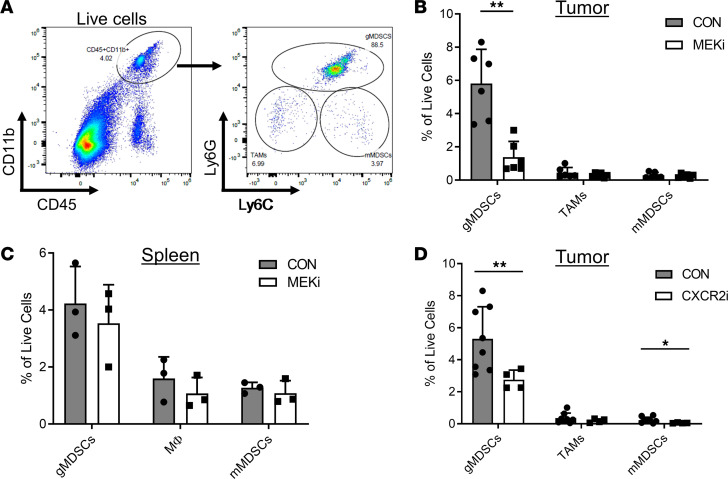
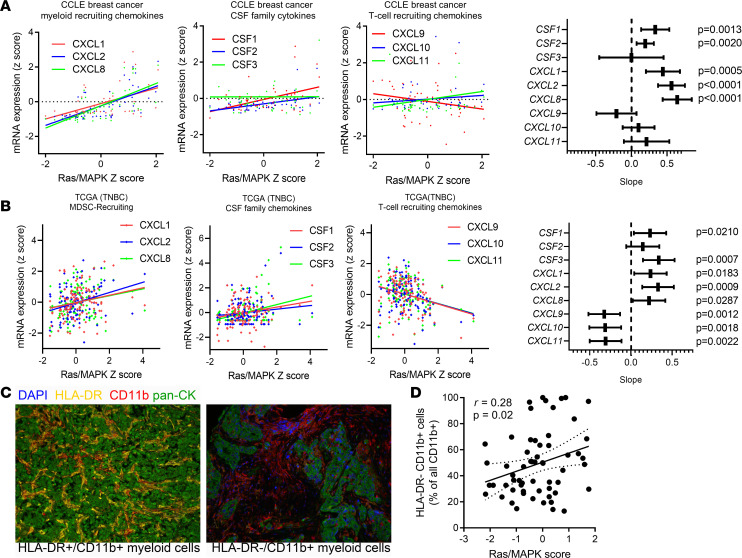
Triple-negative breast cancers (TNBCs) are heterogeneous and aggressive, with high mortality rates. TNBCs frequently respond to chemotherapy, yet many patients develop chemoresistance. The molecular basis and roles for tumor cell-stromal crosstalk in establishing chemoresistance are complex and largely unclear. Here we report molecular studies of paired TNBC patient-derived xenografts (PDXs) established before and after the development of chemoresistance. Interestingly, the chemoresistant model acquired a distinct KRASQ61R mutation that activates K-Ras. The chemoresistant KRAS-mutant model showed gene expression and proteomic changes indicative of altered tumor cell metabolism. Specifically, KRAS-mutant PDXs exhibited increased redox ratios and decreased activation of AMPK, a protein involved in responding to metabolic homeostasis. Additionally, the chemoresistant model exhibited increased immunosuppression, including expression of CXCL1 and CXCL2, cytokines responsible for recruiting immunosuppressive leukocytes to tumors. Notably, chemoresistant KRAS-mutant tumors harbored increased numbers of granulocytic myeloid-derived suppressor cells (gMDSCs). Interestingly, previously established Ras/MAPK-associated gene expression signatures correlated with myeloid/neutrophil-recruiting CXCL1/2 expression and negatively with T cell-recruiting chemokines (CXCL9/10/11) across patients with TNBC, even in the absence of KRAS mutations. MEK inhibition induced tumor suppression in mice while reversing metabolic and immunosuppressive phenotypes, including chemokine production and gMDSC tumor recruitment in the chemoresistant KRAS-mutant tumors. These results suggest that Ras/MAPK pathway inhibitors may be effective in some breast cancer patients to reverse Ras/MAPK-driven tumor metabolism and immunosuppression, particularly in the setting of chemoresistance.
Keywords: Breast cancer; Immunology; Oncology.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
