

A Single-Cell Transcriptomics CRISPR-Activation Screen Identifies Epigenetic Regulators of the Zygotic Genome Activation Program
- PMID: 32634384
- PMCID: PMC7383230
- DOI: 10.1016/j.cels.2020.06.004
A Single-Cell Transcriptomics CRISPR-Activation Screen Identifies Epigenetic Regulators of the Zygotic Genome Activation Program
Abstract
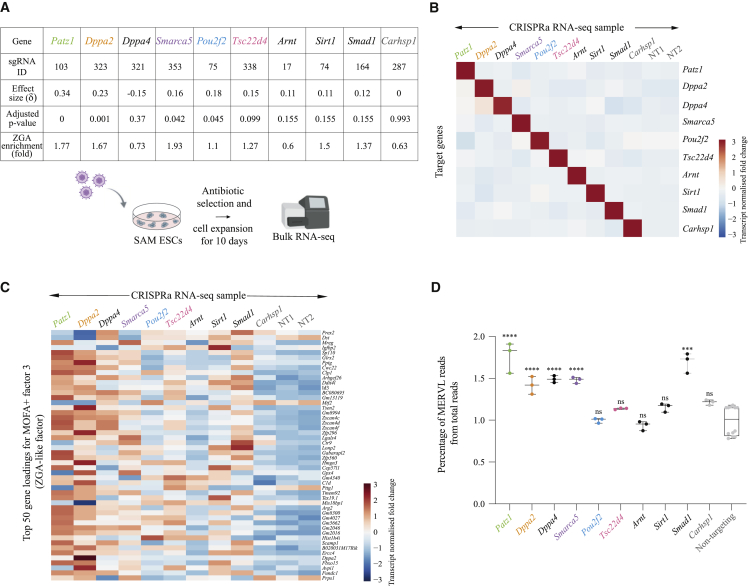
Zygotic genome activation (ZGA) is an essential transcriptional event in embryonic development that coincides with extensive epigenetic reprogramming. Complex manipulation techniques and maternal stores of proteins preclude large-scale functional screens for ZGA regulators within early embryos. Here, we combined pooled CRISPR activation (CRISPRa) with single-cell transcriptomics to identify regulators of ZGA-like transcription in mouse embryonic stem cells, which serve as a tractable, in vitro proxy of early mouse embryos. Using multi-omics factor analysis (MOFA+) applied to ∼200,000 single-cell transcriptomes comprising 230 CRISPRa perturbations, we characterized molecular signatures of ZGA and uncovered 24 factors that promote a ZGA-like response. Follow-up assays validated top screen hits, including the DNA-binding protein Dppa2, the chromatin remodeler Smarca5, and the transcription factor Patz1, and functional experiments revealed that Smarca5's regulation of ZGA-like transcription is dependent on Dppa2. Together, our single-cell transcriptomic profiling of CRISPRa-perturbed cells provides both system-level and molecular insights into the mechanisms that orchestrate ZGA.
Keywords: CRISPRa; Dppa2; MOFA; Patz1; Smarca5; ZGA; scRNA-seq; screen; single cell; zygotic genome activation.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests W.R. is a consultant and shareholder of Cambridge Epigenetix. All other authors declare no competing interests.
Figures







References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
