

Signaling Pathways That Control Muscle Mass
- PMID: 32635462
- PMCID: PMC7369702
- DOI: 10.3390/ijms21134759
Signaling Pathways That Control Muscle Mass
Abstract
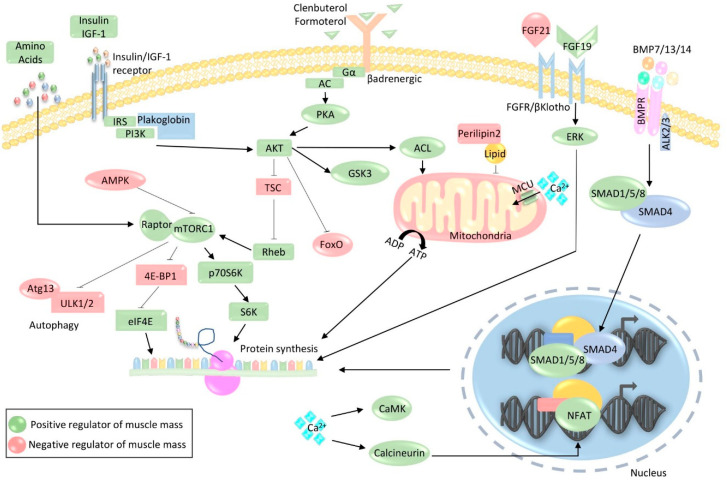
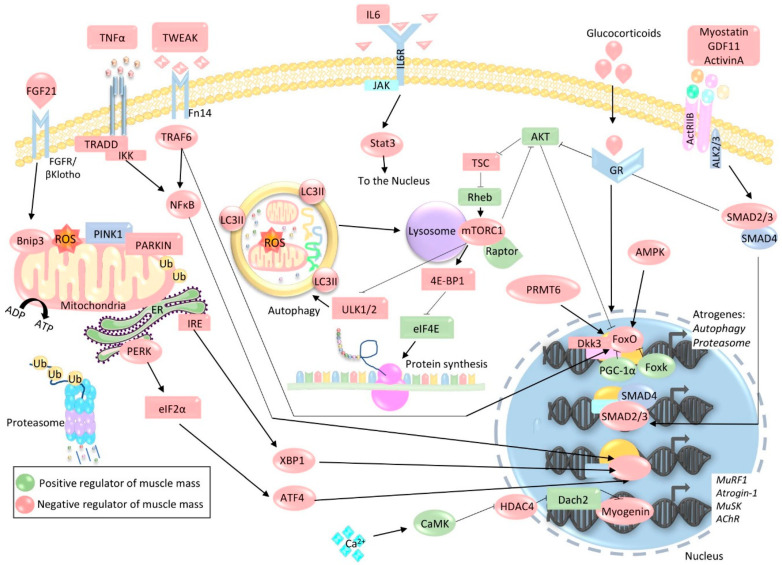
The loss of skeletal muscle mass under a wide range of acute and chronic maladies is associated with poor prognosis, reduced quality of life, and increased mortality. Decades of research indicate the importance of skeletal muscle for whole body metabolism, glucose homeostasis, as well as overall health and wellbeing. This tissue's remarkable ability to rapidly and effectively adapt to changing environmental cues is a double-edged sword. Physiological adaptations that are beneficial throughout life become maladaptive during atrophic conditions. The atrophic program can be activated by mechanical, oxidative, and energetic distress, and is influenced by the availability of nutrients, growth factors, and cytokines. Largely governed by a transcription-dependent mechanism, this program impinges on multiple protein networks including various organelles as well as biosynthetic and quality control systems. Although modulating muscle function to prevent and treat disease is an enticing concept that has intrigued research teams for decades, a lack of thorough understanding of the molecular mechanisms and signaling pathways that control muscle mass, in addition to poor transferability of findings from rodents to humans, has obstructed efforts to develop effective treatments. Here, we review the progress made in unraveling the molecular mechanisms responsible for the regulation of muscle mass, as this continues to be an intensive area of research.
Keywords: atrophy; hypertrophy; sarcopenia; skeletal muscle.
Conflict of interest statement
Anna Vainshtein is a co-founder of Craft Science Inc., in which she holds shares. The sponsors had no role in the writing of this article or the decision to submit it for publication.
Figures
Similar articles
-
Protein kinase B/Akt: a nexus of growth factor and cytokine signaling in determining muscle mass.J Appl Physiol (1985). 2007 Jul;103(1):378-87. doi: 10.1152/japplphysiol.00089.2007. Epub 2007 Mar 1. J Appl Physiol (1985). 2007. PMID: 17332274 Review.
-
Mechanisms of muscle atrophy and hypertrophy: implications in health and disease.Nat Commun. 2021 Jan 12;12(1):330. doi: 10.1038/s41467-020-20123-1. Nat Commun. 2021. PMID: 33436614 Free PMC article. Review.
-
Molecular mechanisms modulating muscle mass.Trends Mol Med. 2003 Aug;9(8):344-50. doi: 10.1016/s1471-4914(03)00138-2. Trends Mol Med. 2003. PMID: 12928036 Review.
-
Skeletal muscle: increasing the size of the locomotor cell.Int J Biochem Cell Biol. 2010 Sep;42(9):1376-9. doi: 10.1016/j.biocel.2010.05.013. Epub 2010 Jun 9. Int J Biochem Cell Biol. 2010. PMID: 20541033 Review.
-
Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword?FEBS J. 2013 Sep;280(17):4131-48. doi: 10.1111/febs.12338. Epub 2013 Jun 18. FEBS J. 2013. PMID: 23663276 Free PMC article. Review.
Cited by
-
Protein Extraction Methods Suitable for Muscle Tissue Proteomic Analysis.Proteomes. 2024 Sep 25;12(4):27. doi: 10.3390/proteomes12040027. Proteomes. 2024. PMID: 39449499 Free PMC article.
-
Mechanisms of exercise as a preventative measure to muscle wasting.Am J Physiol Cell Physiol. 2021 Jul 1;321(1):C40-C57. doi: 10.1152/ajpcell.00056.2021. Epub 2021 May 5. Am J Physiol Cell Physiol. 2021. PMID: 33950699 Free PMC article. Review.
-
Endurance Exercise-Induced Fgf21 Promotes Skeletal Muscle Fiber Conversion through TGF-β1 and p38 MAPK Signaling Pathway.Int J Mol Sci. 2023 Jul 13;24(14):11401. doi: 10.3390/ijms241411401. Int J Mol Sci. 2023. PMID: 37511159 Free PMC article.
-
Skeletal Muscle Recovery from Disuse Atrophy: Protein Turnover Signaling and Strategies for Accelerating Muscle Regrowth.Int J Mol Sci. 2020 Oct 26;21(21):7940. doi: 10.3390/ijms21217940. Int J Mol Sci. 2020. PMID: 33114683 Free PMC article. Review.
-
Inflammatory Markers Associated with Physical Frailty and Cognitive Impairment.Aging Dis. 2024 Apr 17;16(2):859-875. doi: 10.14336/AD.2024.0258. Aging Dis. 2024. PMID: 38739942 Free PMC article. Review.
References
-
- Tang H., Inoki K., Lee M., Wright E., Khuong A.A., Khuong A.A., Sugiarto S., Garner M., Paik J., DePinho R.A., et al. mTORC1 promotes denervation-induced muscle atrophy through a mechanism involving the activation of FoxO and E3 ubiquitin ligases. Sci. Signal. 2014;7:ra18. doi: 10.1126/scisignal.2004809. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
