

Autophagy Roles in Genome Maintenance
- PMID: 32635505
- PMCID: PMC7407194
- DOI: 10.3390/cancers12071793
Autophagy Roles in Genome Maintenance
Abstract
In recent years, a considerable correlation has emerged between autophagy and genome integrity. A range of mechanisms appear to be involved where autophagy participates in preventing genomic instability, as well as in DNA damage response and cell fate decision. These initial findings have attracted particular attention in the context of malignancy; however, the crosstalk between autophagy and DNA damage response is just beginning to be explored and key questions remain that need to be addressed, to move this area of research forward and illuminate the overall consequence of targeting this process in human therapies. Here we present current knowledge on the complex crosstalk between autophagy and genome integrity and discuss its implications for cancer cell survival and response to therapy.
Keywords: DNA damage repair; DNA damage response; autophagy; cancer therapy.
Conflict of interest statement
No potential conflicts of interest were disclosed.
Figures
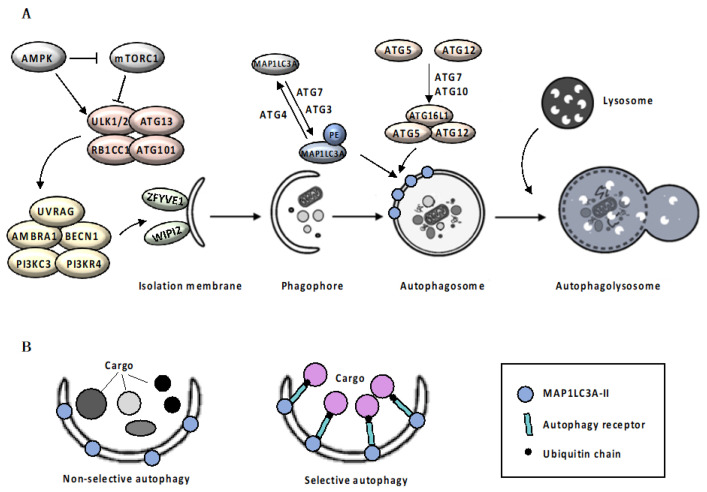

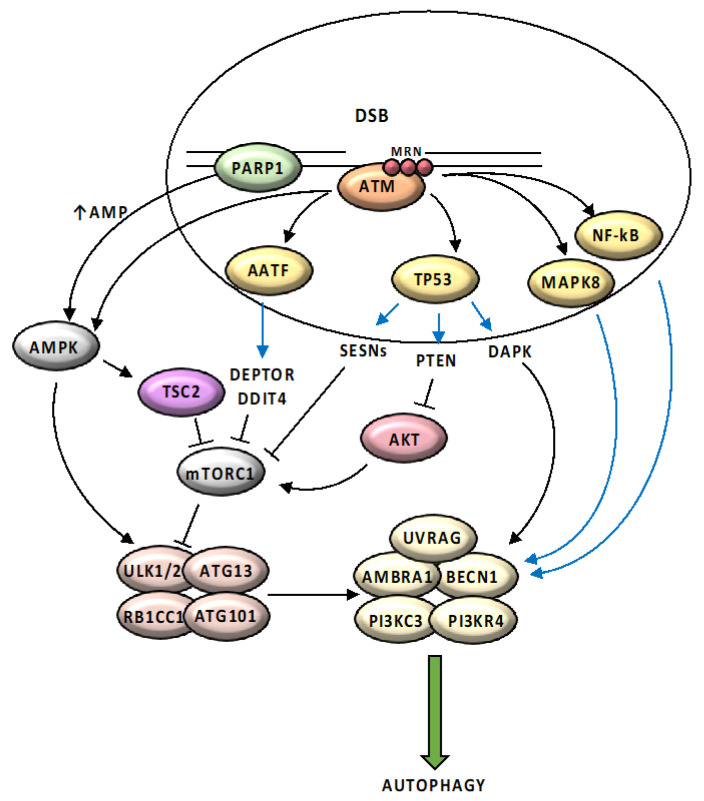
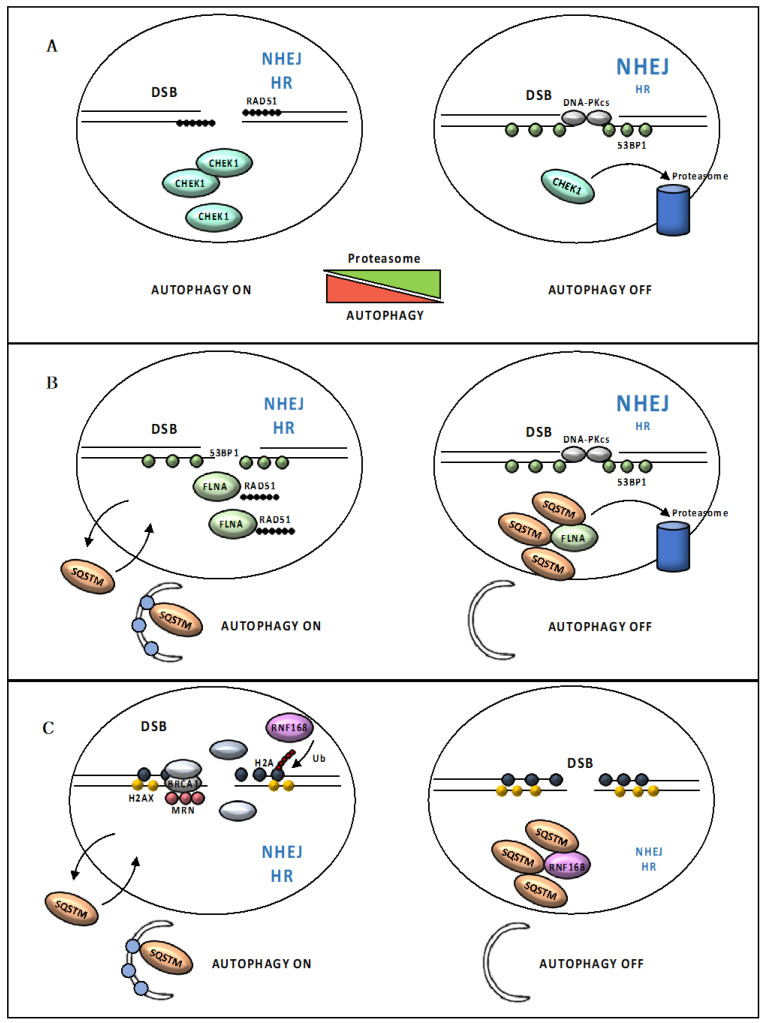


References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
