

Single-cell ATAC sequencing analysis: From data preprocessing to hypothesis generation
- PMID: 32637041
- PMCID: PMC7327298
- DOI: 10.1016/j.csbj.2020.06.012
Single-cell ATAC sequencing analysis: From data preprocessing to hypothesis generation
Abstract
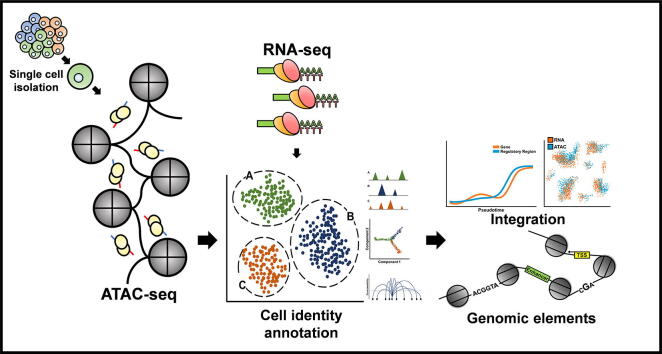
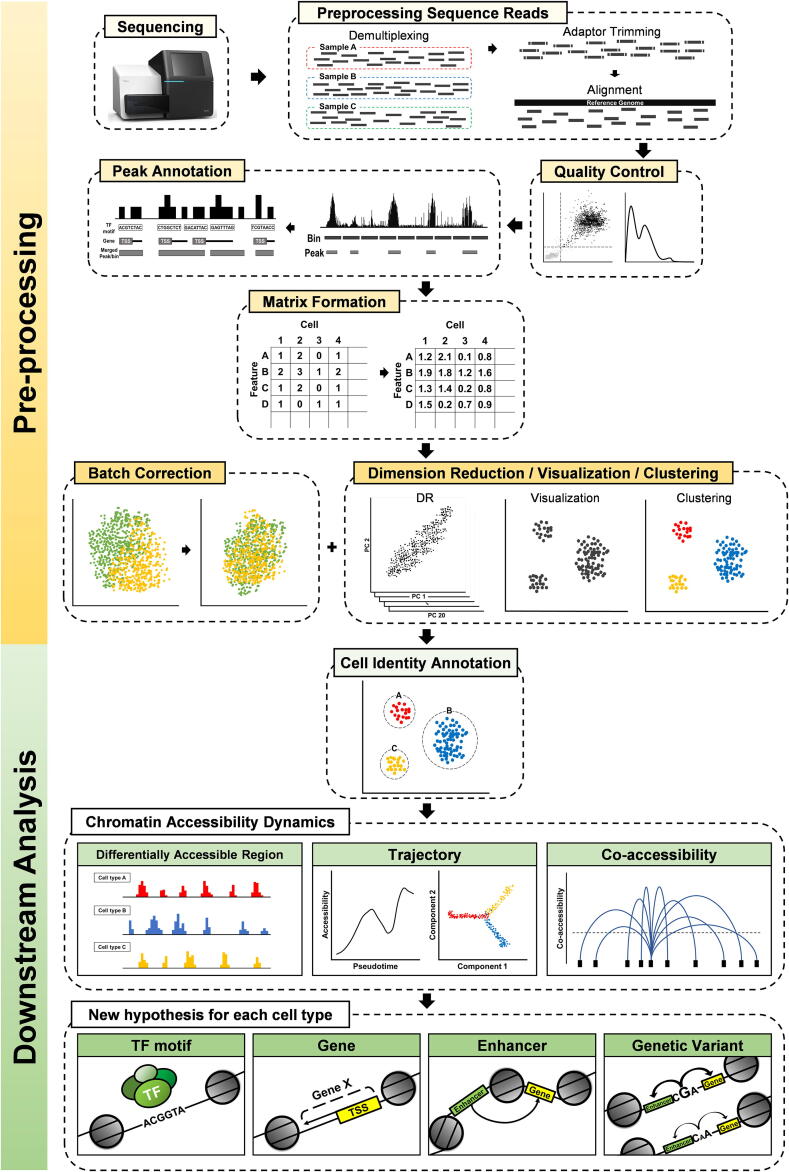
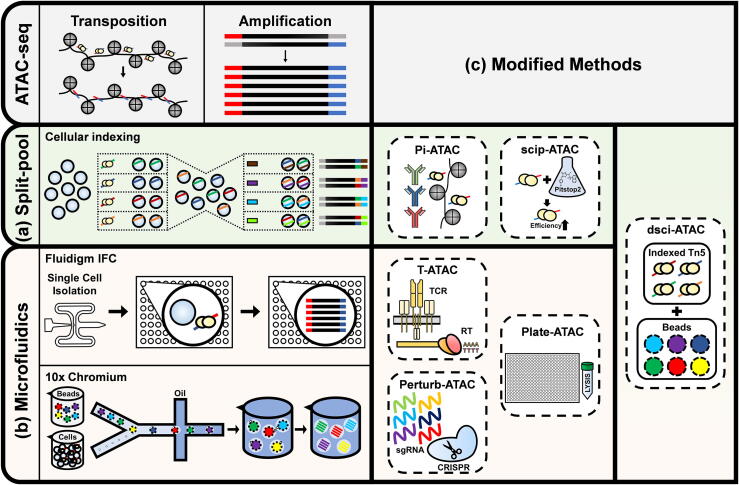
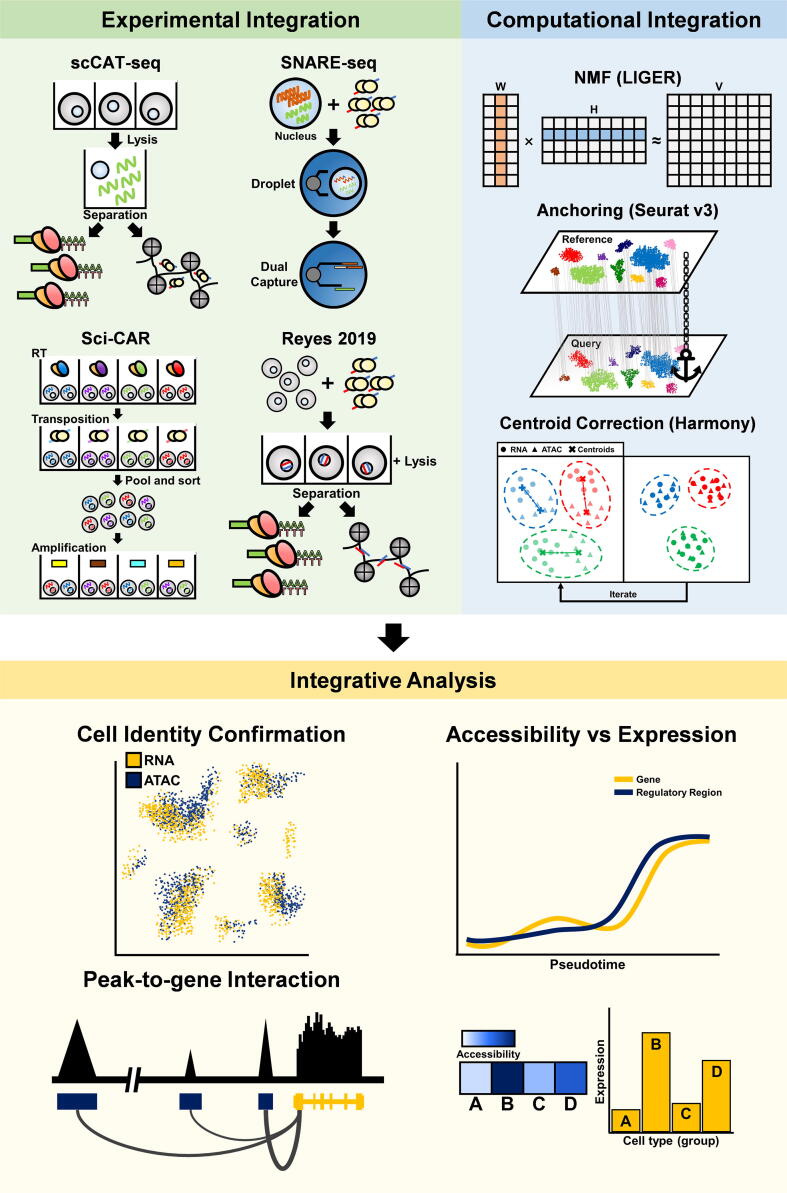
Most genetic variations associated with human complex traits are located in non-coding genomic regions. Therefore, understanding the genotype-to-phenotype axis requires a comprehensive catalog of functional non-coding genomic elements, most of which are involved in epigenetic regulation of gene expression. Genome-wide maps of open chromatin regions can facilitate functional analysis of cis- and trans-regulatory elements via their connections with trait-associated sequence variants. Currently, Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq) is considered the most accessible and cost-effective strategy for genome-wide profiling of chromatin accessibility. Single-cell ATAC-seq (scATAC-seq) technology has also been developed to study cell type-specific chromatin accessibility in tissue samples containing a heterogeneous cellular population. However, due to the intrinsic nature of scATAC-seq data, which are highly noisy and sparse, accurate extraction of biological signals and devising effective biological hypothesis are difficult. To overcome such limitations in scATAC-seq data analysis, new methods and software tools have been developed over the past few years. Nevertheless, there is no consensus for the best practice of scATAC-seq data analysis yet. In this review, we discuss scATAC-seq technology and data analysis methods, ranging from preprocessing to downstream analysis, along with an up-to-date list of published studies that involved the application of this method. We expect this review will provide a guideline for successful data generation and analysis methods using appropriate software tools and databases for the study of chromatin accessibility at single-cell resolution.
Keywords: ATAC sequencing; Chromatin accessibility; Single-cell ATAC sequencing; Single-cell RNA sequencing; Single-cell biology.
© 2020 The Author(s).
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous