

Handling multi-mapped reads in RNA-seq
- PMID: 32637053
- PMCID: PMC7330433
- DOI: 10.1016/j.csbj.2020.06.014
Handling multi-mapped reads in RNA-seq
Abstract
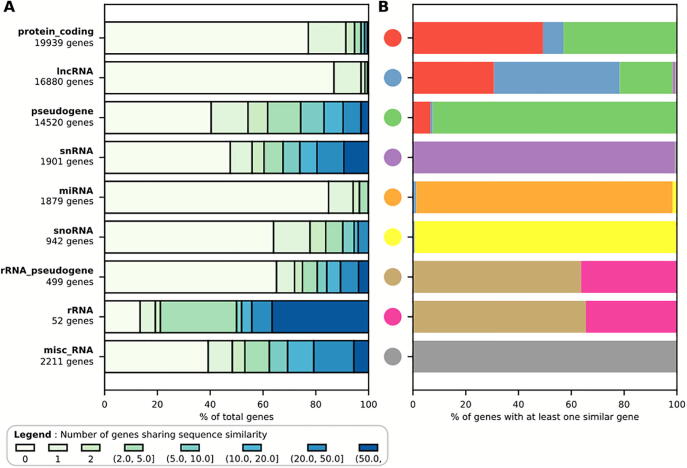
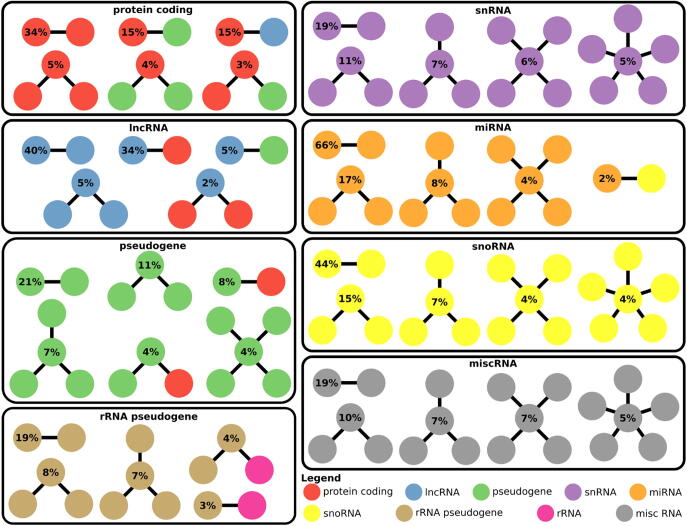
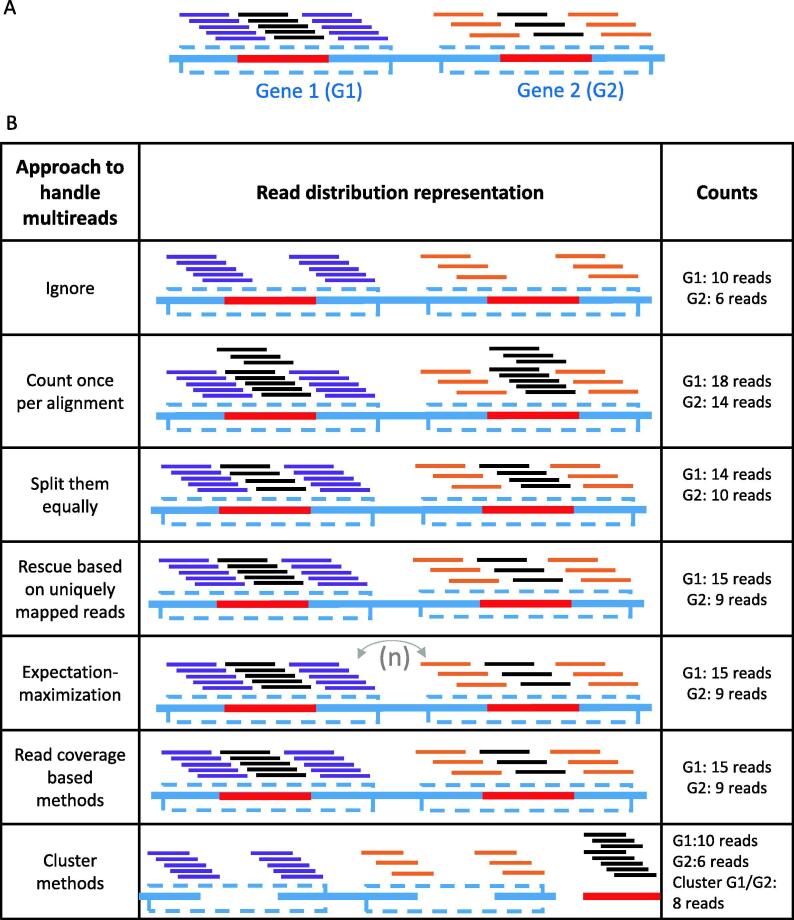
Many eukaryotic genomes harbour large numbers of duplicated sequences, of diverse biotypes, resulting from several mechanisms including recombination, whole genome duplication and retro-transposition. Such repeated sequences complicate gene/transcript quantification during RNA-seq analysis due to reads mapping to more than one locus, sometimes involving genes embedded in other genes. Genes of different biotypes have dissimilar levels of sequence duplication, with long-noncoding RNAs and messenger RNAs sharing less sequence similarity to other genes than biotypes encoding shorter RNAs. Many strategies have been elaborated to handle these multi-mapped reads, resulting in increased accuracy in gene/transcript quantification, although separate tools are typically used to estimate the abundance of short and long genes due to their dissimilar characteristics. This review discusses the mechanisms leading to sequence duplication, the biotypes affected, the computational strategies employed to deal with multi-mapped reads and the challenges that still remain to be overcome.
Keywords: Duplicated genes; Expectation–maximization algorithm; Gene isoforms; Multi-mapped reads; Noncoding RNAs; RNA-seq.
© 2020 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Ohta T. Role of gene duplication in evolution. Genome. 1989;31:304–310. - PubMed
-
- Magadum S., Banerjee U., Murugan P., Gangapur D., Ravikesavan R. Gene duplication as a major force in evolution. J Genet. 2013;92:155–161. - PubMed
-
- Dharshini S.A.P., Taguchi Y.H., Gromiha M.M. Identifying suitable tools for variant detection and differential gene expression using RNA-seq data. Genomics. 2020;112:2166–2172. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources