

Site directed mutagenesis as a precision tool to enable synthetic biology with engineered modular polyketide synthases
- PMID: 32637664
- PMCID: PMC7327777
- DOI: 10.1016/j.synbio.2020.04.001
Site directed mutagenesis as a precision tool to enable synthetic biology with engineered modular polyketide synthases
Erratum in
-
Corrigendum to "Site directed mutagenesis as a precision tool to enable synthetic biology with engineered modular polyketide synthases" [Synth Syst Biotechnol 5 (2) (2020) 62-80].Synth Syst Biotechnol. 2020 Aug 6;5(4):268. doi: 10.1016/j.synbio.2020.07.007. eCollection 2020 Dec. Synth Syst Biotechnol. 2020. PMID: 32817884 Free PMC article.
-
Erratum regarding previously published articles.Synth Syst Biotechnol. 2020 Oct 14;5(4):332. doi: 10.1016/j.synbio.2020.10.004. eCollection 2020 Dec. Synth Syst Biotechnol. 2020. PMID: 33102828 Free PMC article.
Abstract
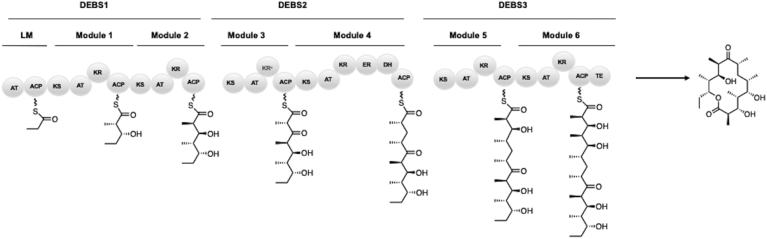
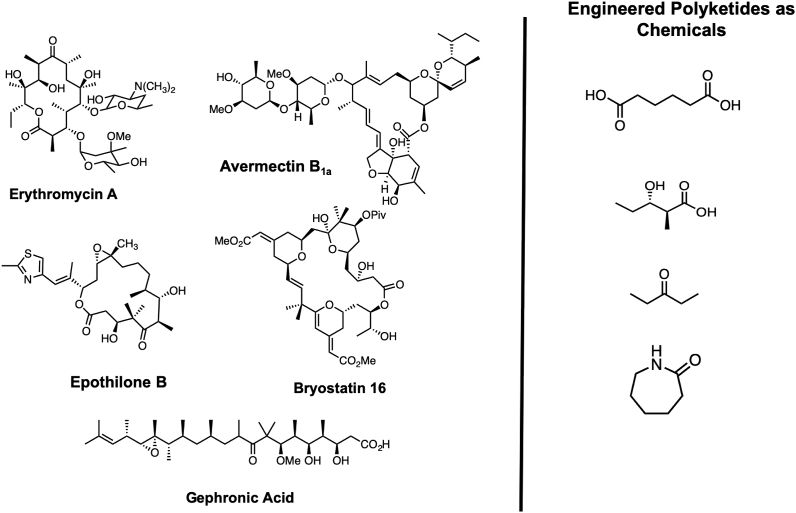
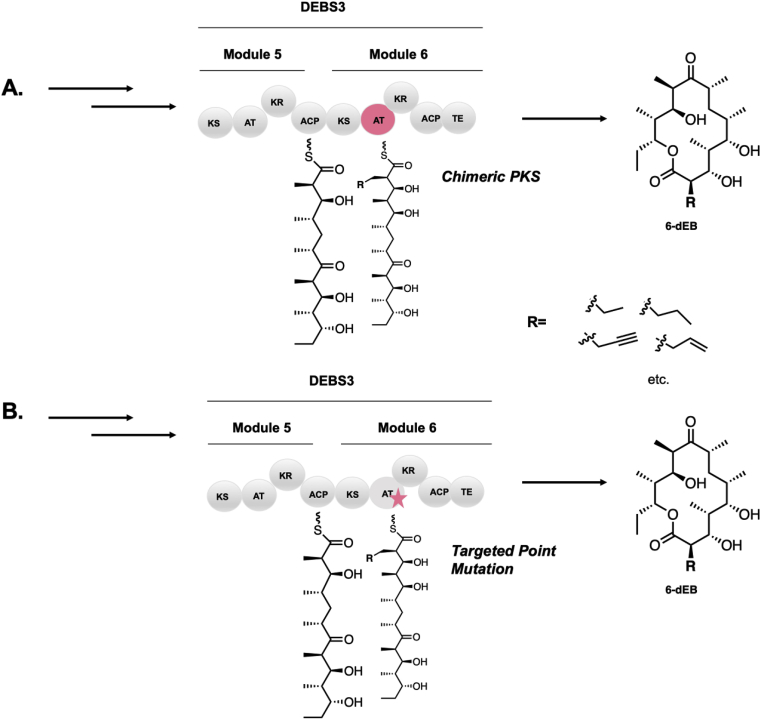
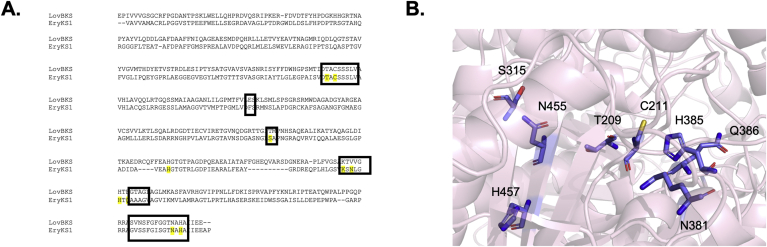
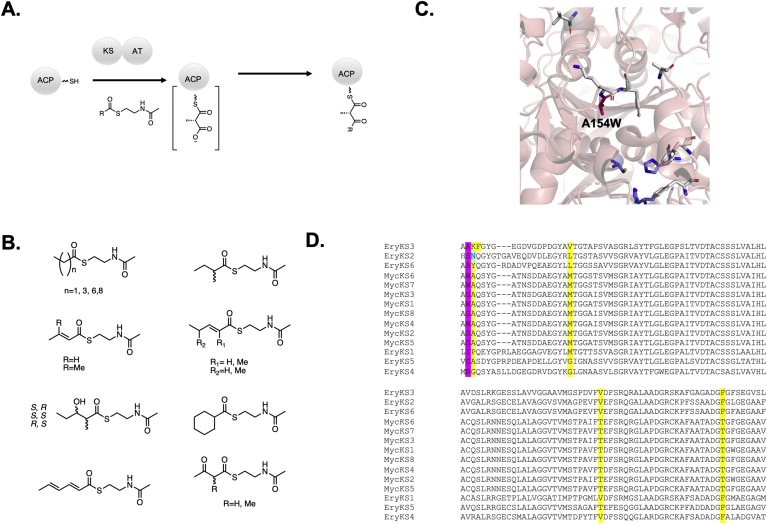
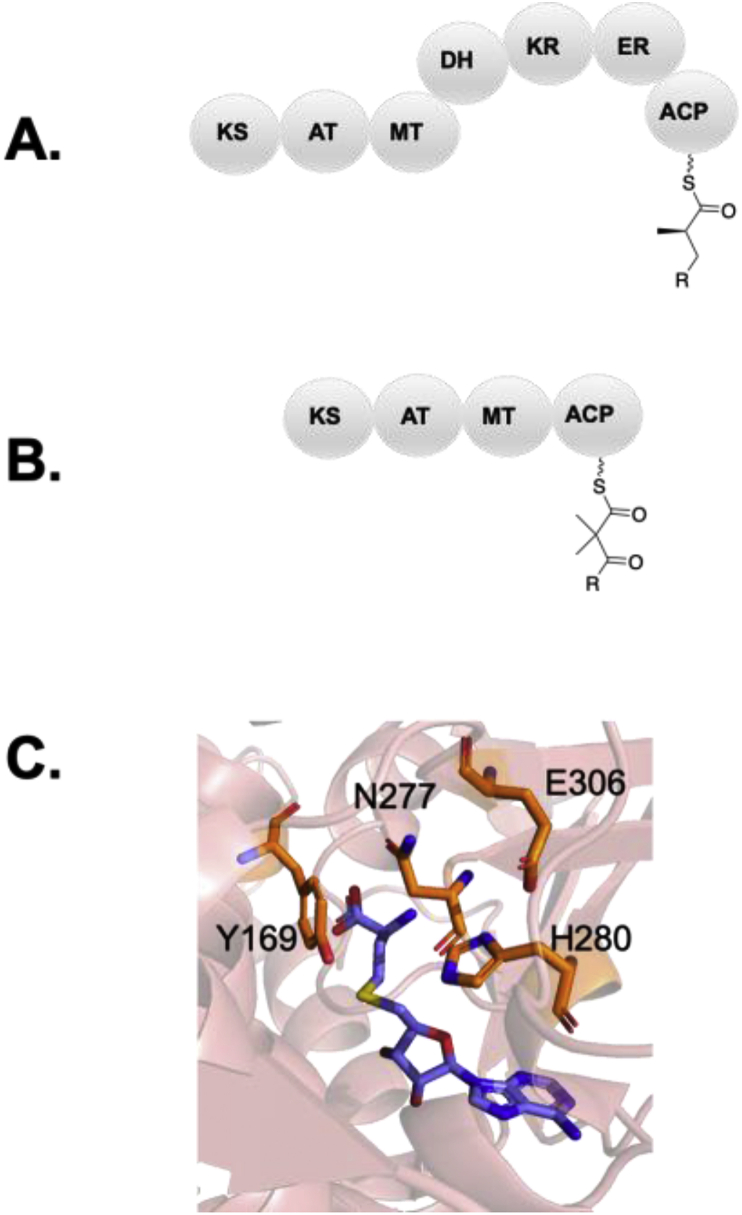
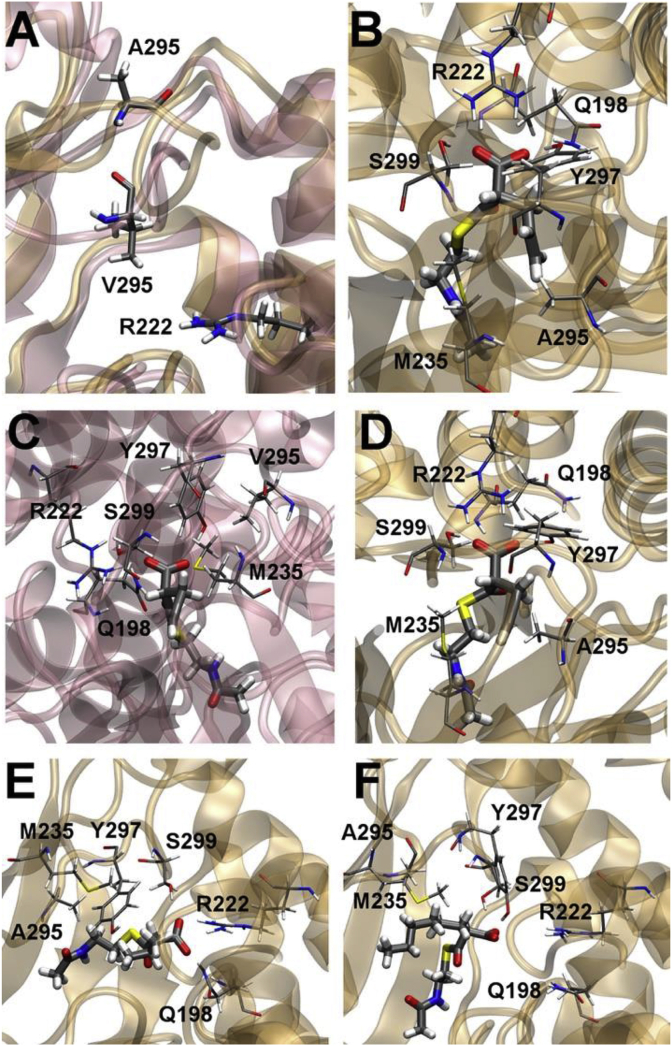
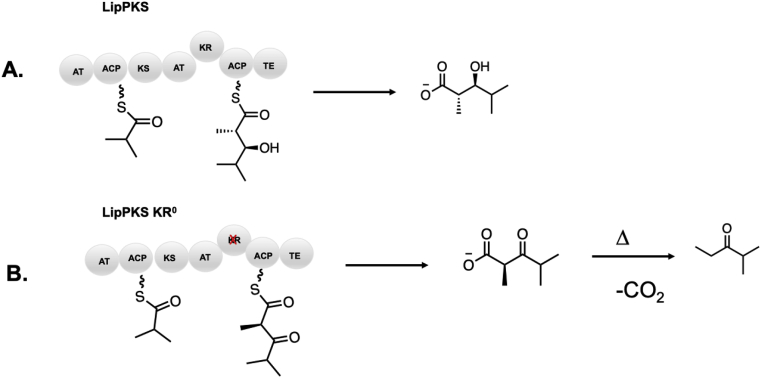
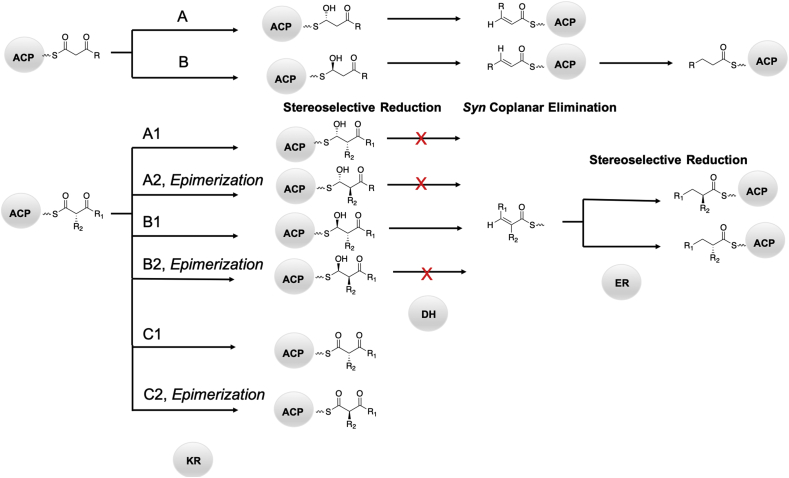
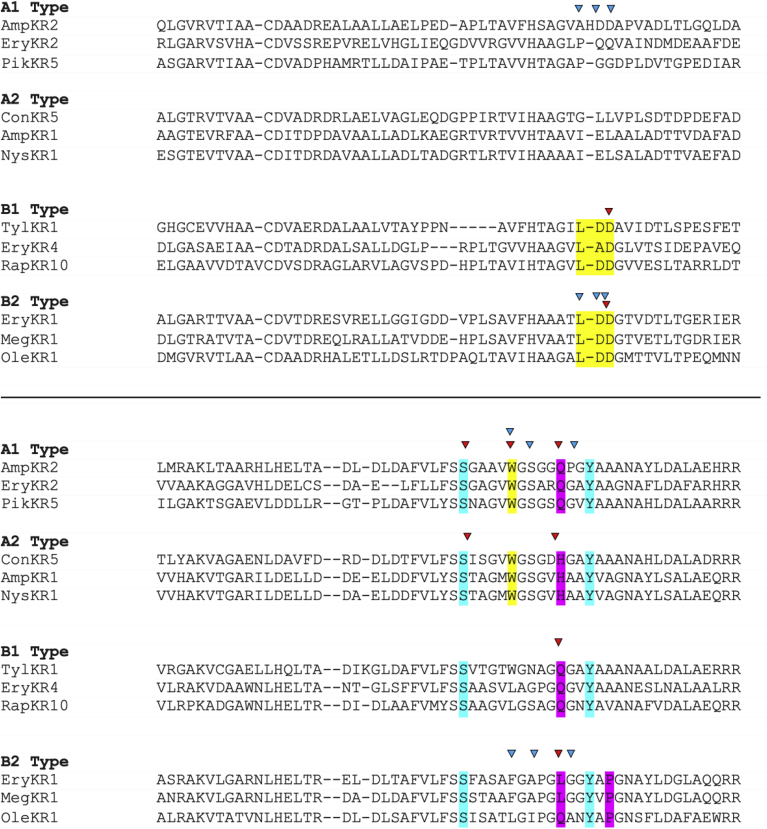
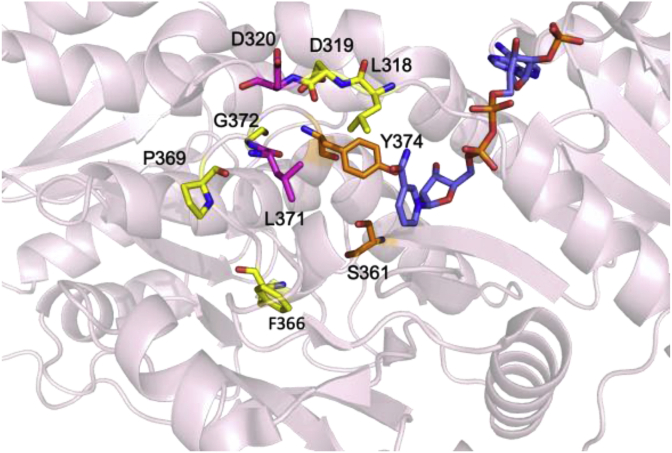
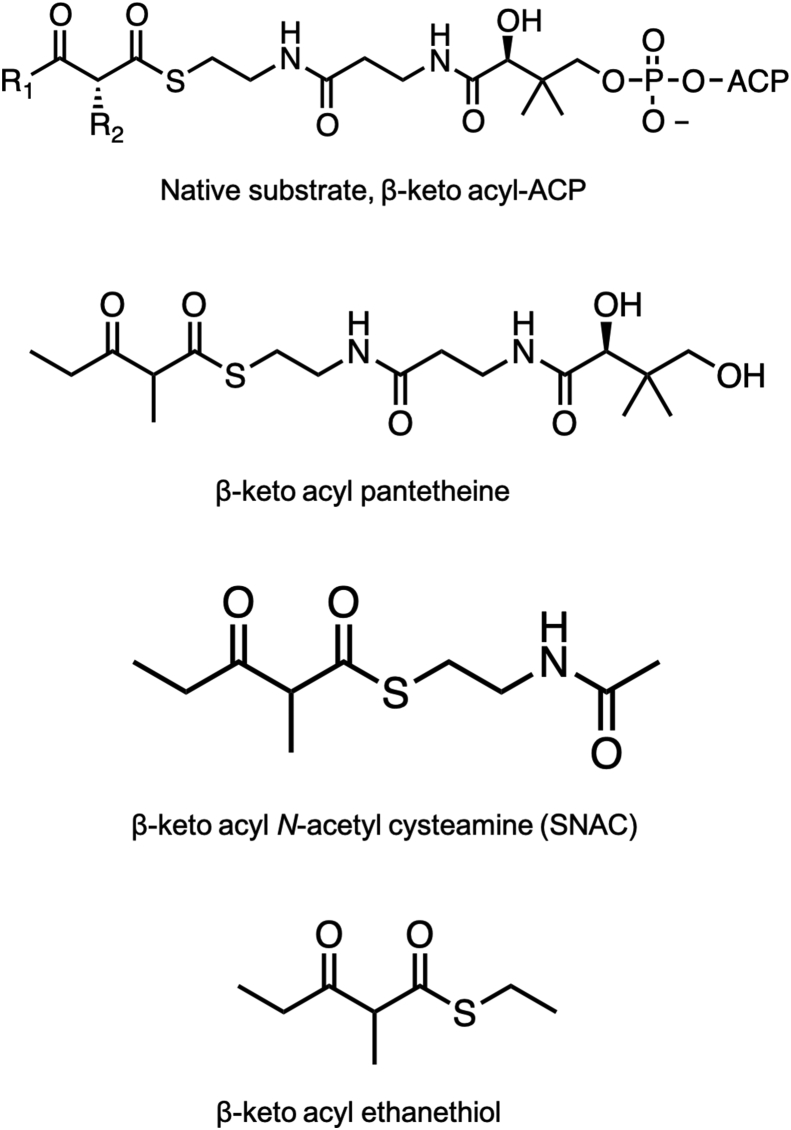
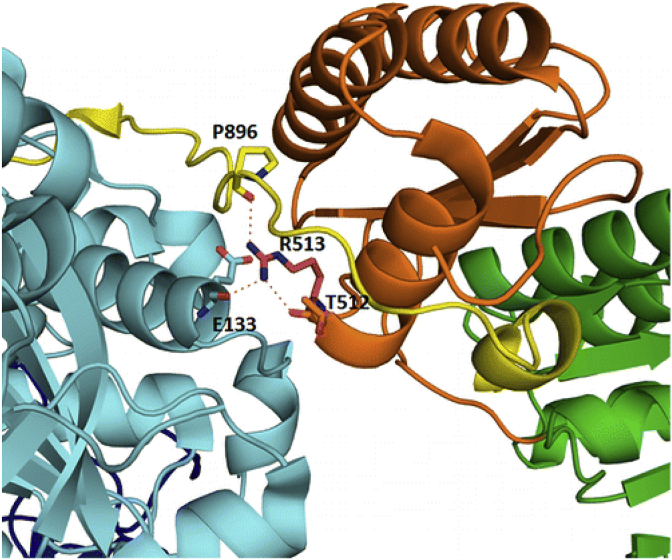
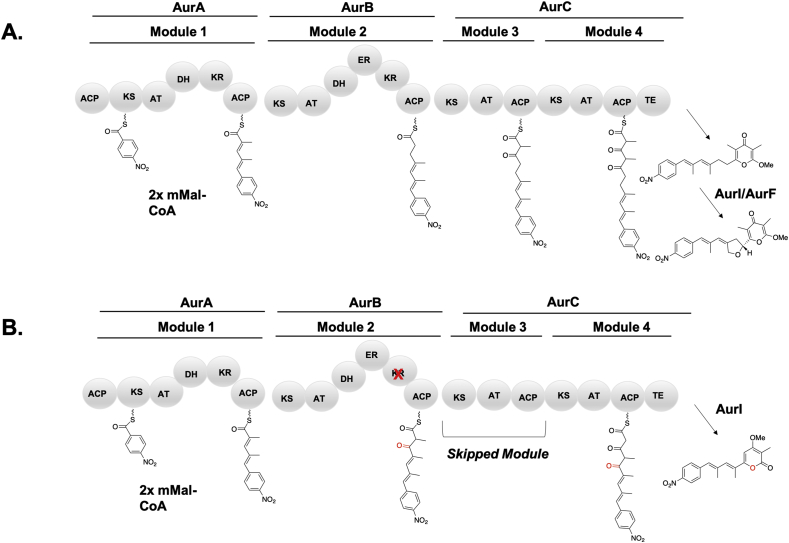
Modular polyketide synthases (PKSs) are a multidomain megasynthase class of biosynthetic enzymes that have great promise for the development of new compounds, from new pharmaceuticals to high value commodity and specialty chemicals. Their colinear biosynthetic logic has been viewed as a promising platform for synthetic biology for decades. Due to this colinearity, domain swapping has long been used as a strategy to introduce molecular diversity. However, domain swapping often fails because it perturbs critical protein-protein interactions within the PKS. With our increased level of structural elucidation of PKSs, using judicious targeted mutations of individual residues is a more precise way to introduce molecular diversity with less potential for global disruption of the protein architecture. Here we review examples of targeted point mutagenesis to one or a few residues harbored within the PKS that alter domain specificity or selectivity, affect protein stability and interdomain communication, and promote more complex catalytic reactivity.
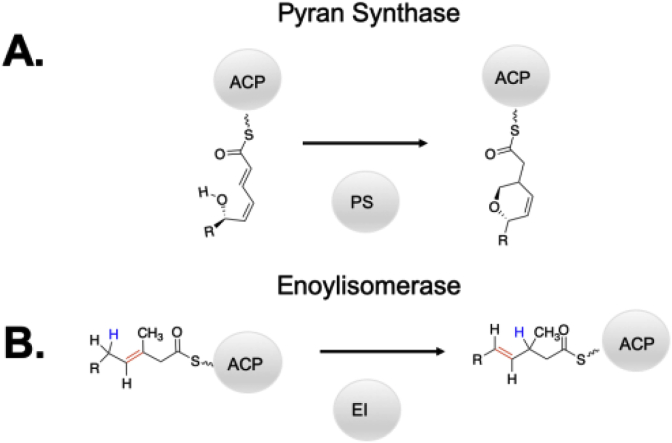
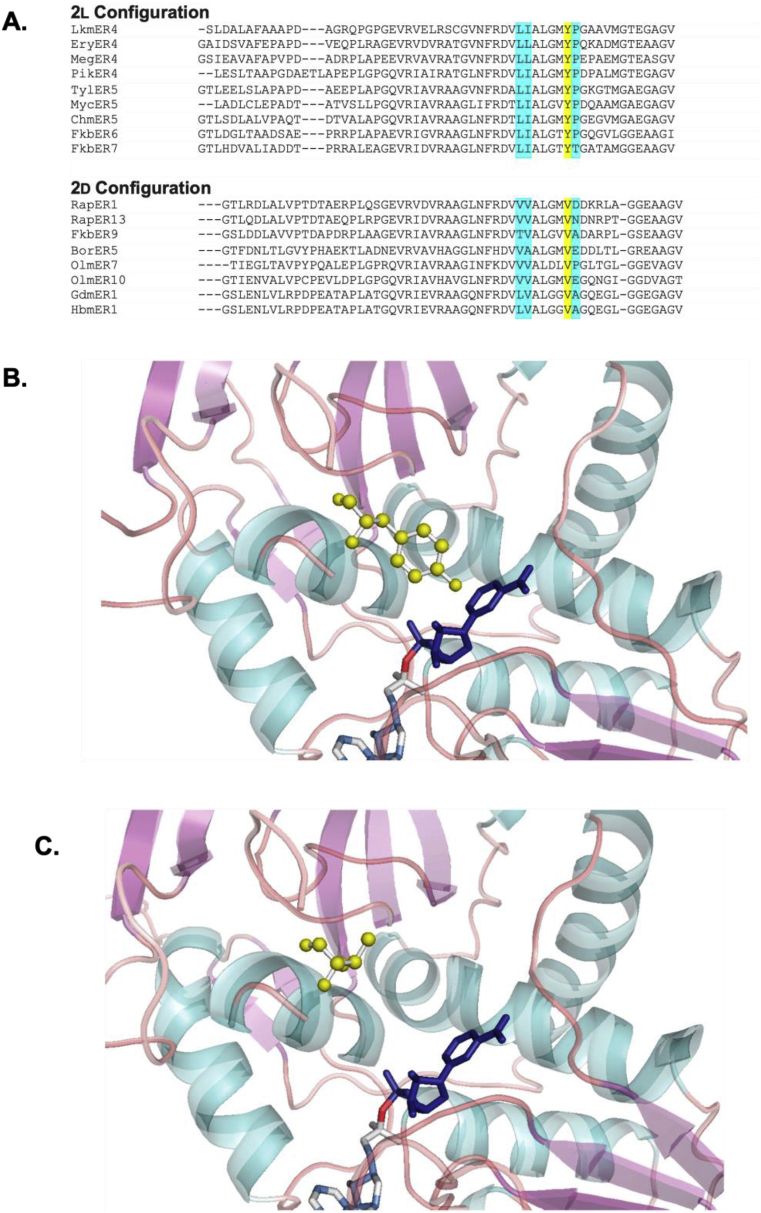
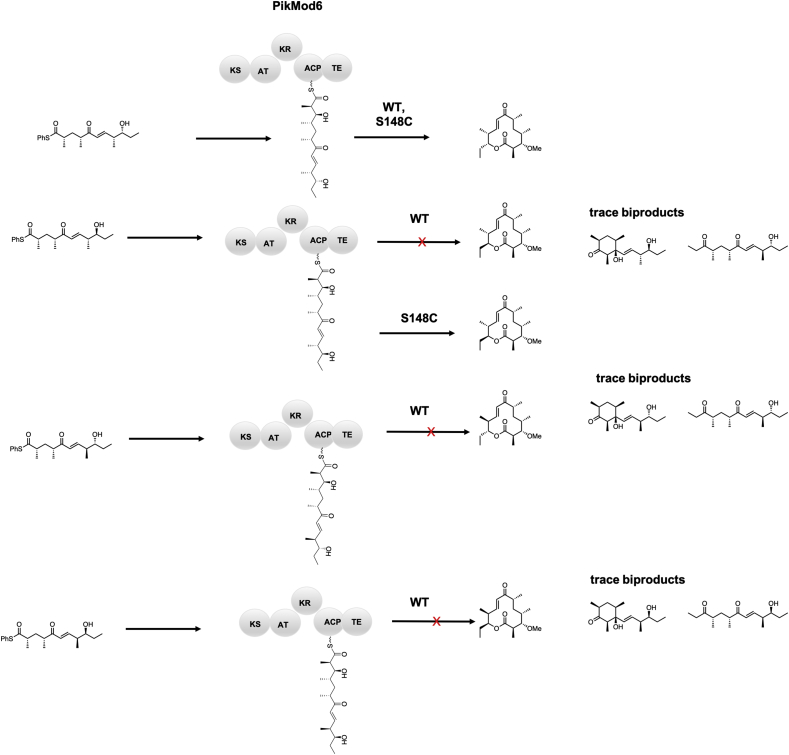
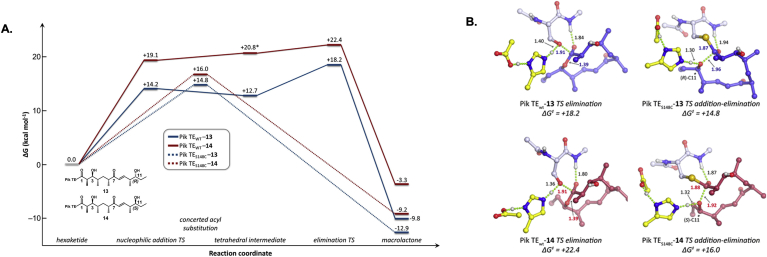
Keywords: ACP, acyl carrier protein; AT, acyltransferase; DEBS, 6-deoxyerthronolide B synthase; DH, dehydratase; EI, enoylisomerase; ER, enoylreductase; KR, ketoreductase; KS, ketosynthase; LM, loading module; MT, methyltransferase; Mod, module; PKS, polyketide synthase; PS, pyran synthase; Polyketide synthase; Protein engineering; Rational design; SNAC, N-acetyl cysteamine; Saturation mutagenesis; Site directed mutagenesis; Synthetic biology.
© 2020 Production and hosting by Elsevier B.V. on behalf of KeAi Communications Co., Ltd.
Figures

References
-
- Yuzawa S. Commodity chemicals from engineered modular type I polyketide synthases. 2018;608 - PubMed
-
- Hagen A. Engineering a polyketide synthase for in vitro production of adipic acid. ACS Synth Biol. 2016;5:21–27. - PubMed
-
- Menzella H.G. Rational design and assembly of synthetic trimodular polyketide synthases. Chem Biol. 2007;14:143–151. - PubMed
-
- Donadio S. Modular organization of genes required for complex polyketide biosynthesis. Science. 1991;252:675–679. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
