

Leveraging Systematic Functional Analysis to Benchmark an In Silico Framework Distinguishes Driver from Passenger MEK Mutants in Cancer
- PMID: 32641410
- PMCID: PMC7541597
- DOI: 10.1158/0008-5472.CAN-20-0865
Leveraging Systematic Functional Analysis to Benchmark an In Silico Framework Distinguishes Driver from Passenger MEK Mutants in Cancer
Abstract
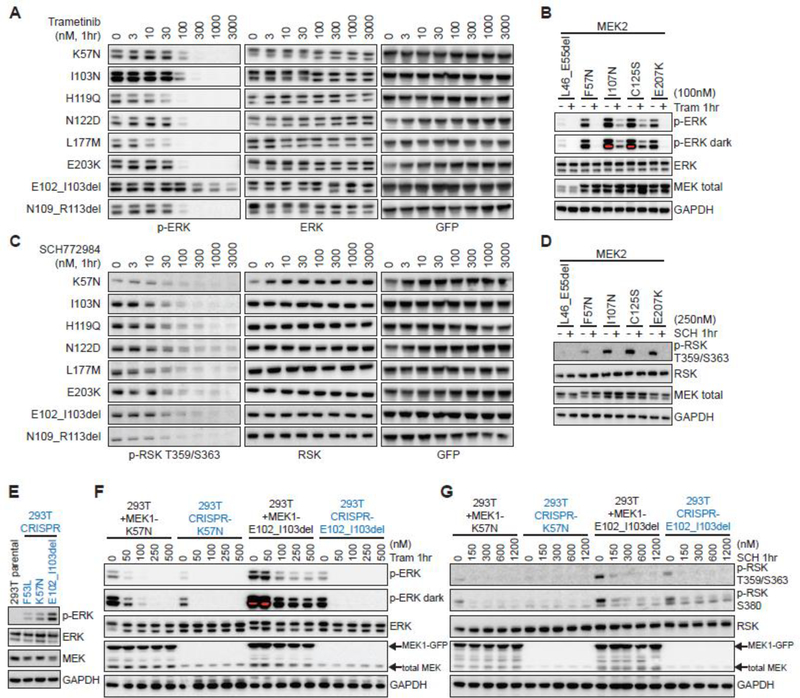
Despite significant advances in cancer precision medicine, a significant hurdle to its broader adoption remains the multitude of variants of unknown significance identified by clinical tumor sequencing and the lack of biologically validated methods to distinguish between functional and benign variants. Here we used functional data on MAP2K1 and MAP2K2 mutations generated in real-time within a co-clinical trial framework to benchmark the predictive value of a three-part in silico methodology. Our computational approach to variant classification incorporated hotspot analysis, three-dimensional molecular dynamics simulation, and sequence paralogy. In silico prediction accurately distinguished functional from benign MAP2K1 and MAP2K2 mutants, yet drug sensitivity varied widely among activating mutant alleles. These results suggest that multifaceted in silico modeling can inform patient accrual to MEK/ERK inhibitor clinical trials, but computational methods need to be paired with laboratory- and clinic-based efforts designed to unravel variabilities in drug response. SIGNIFICANCE: Leveraging prospective functional characterization of MEK1/2 mutants, it was found that hotspot analysis, molecular dynamics simulation, and sequence paralogy are complementary tools that can robustly prioritize variants for biologic, therapeutic, and clinical validation.See related commentary by Whitehead and Sebolt-Leopold, p. 4042.
©2020 American Association for Cancer Research.
Figures
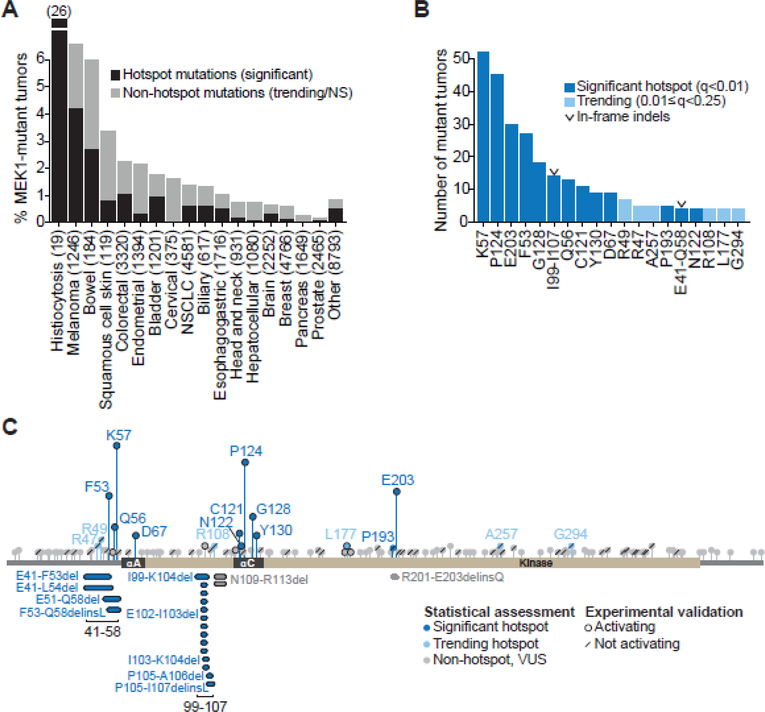
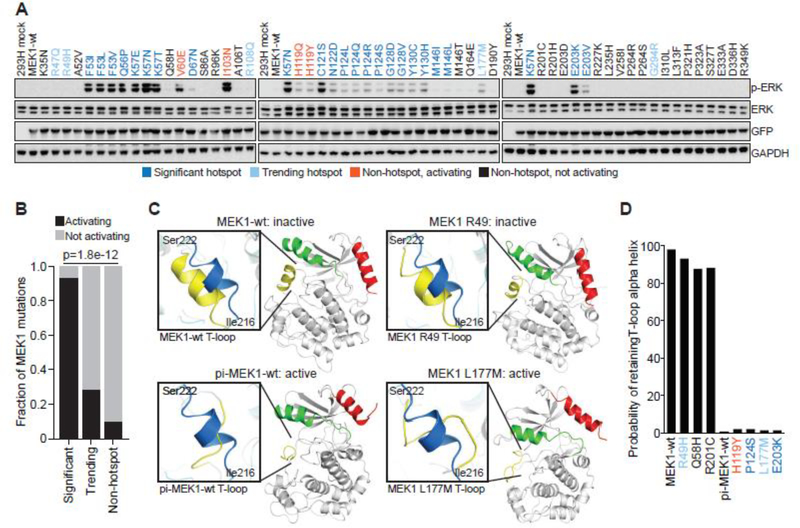
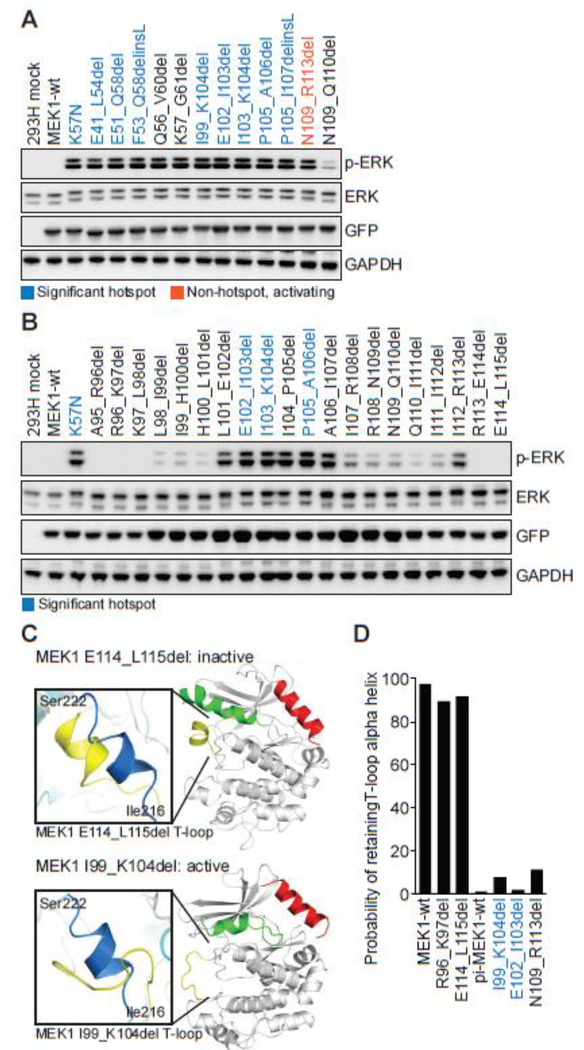
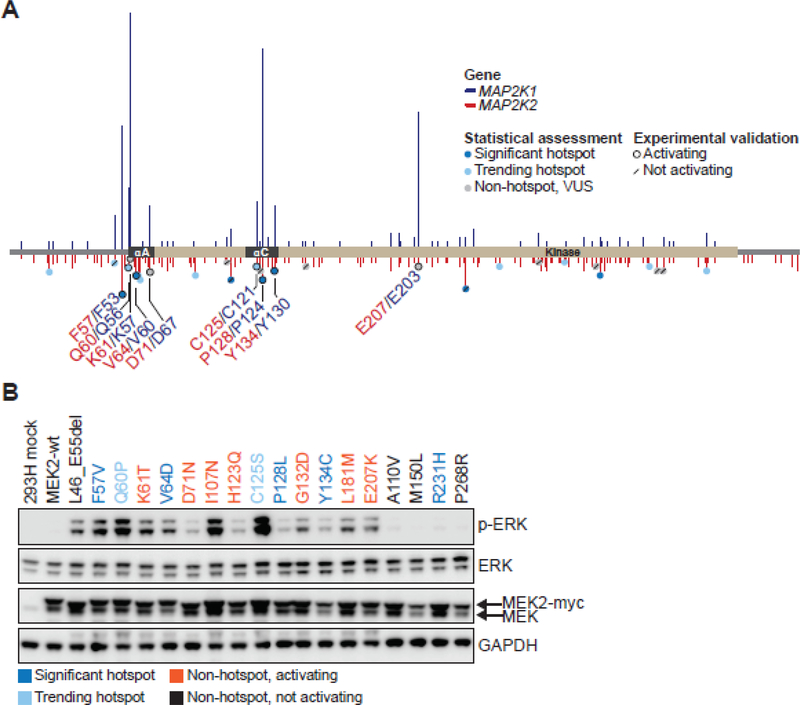
Comment in
-
Deciphering the Complexity of MEK Mutations in the Clinic.Cancer Res. 2020 Oct 1;80(19):4042-4043. doi: 10.1158/0008-5472.CAN-20-2611. Cancer Res. 2020. PMID: 33008803
Comment on
-
Deciphering the Complexity of MEK Mutations in the Clinic.Cancer Res. 2020 Oct 1;80(19):4042-4043. doi: 10.1158/0008-5472.CAN-20-2611. Cancer Res. 2020. PMID: 33008803
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
