

A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing
- PMID: 32641830
- PMCID: PMC7381381
- DOI: 10.1038/s41586-020-2477-4
A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing
Abstract
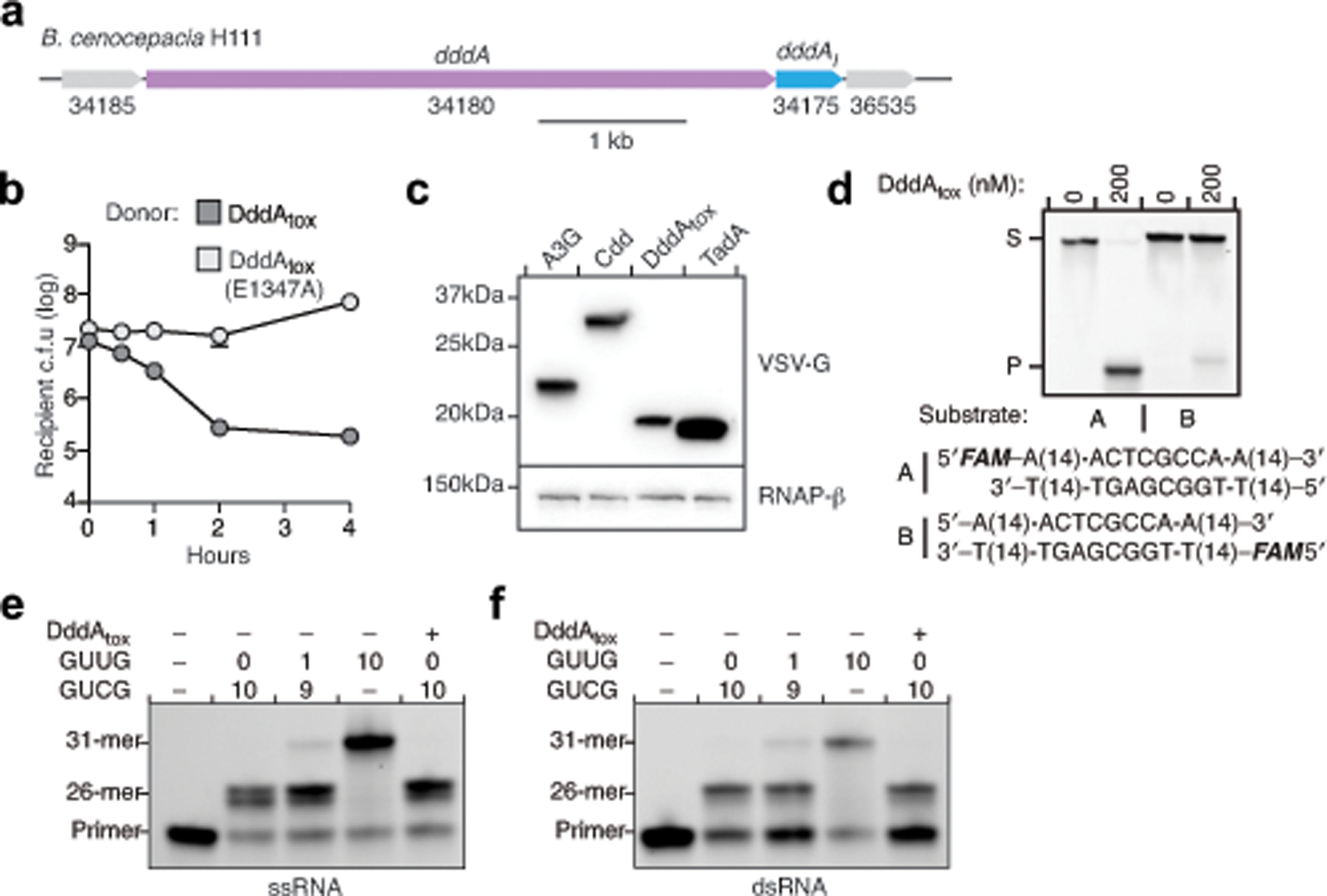
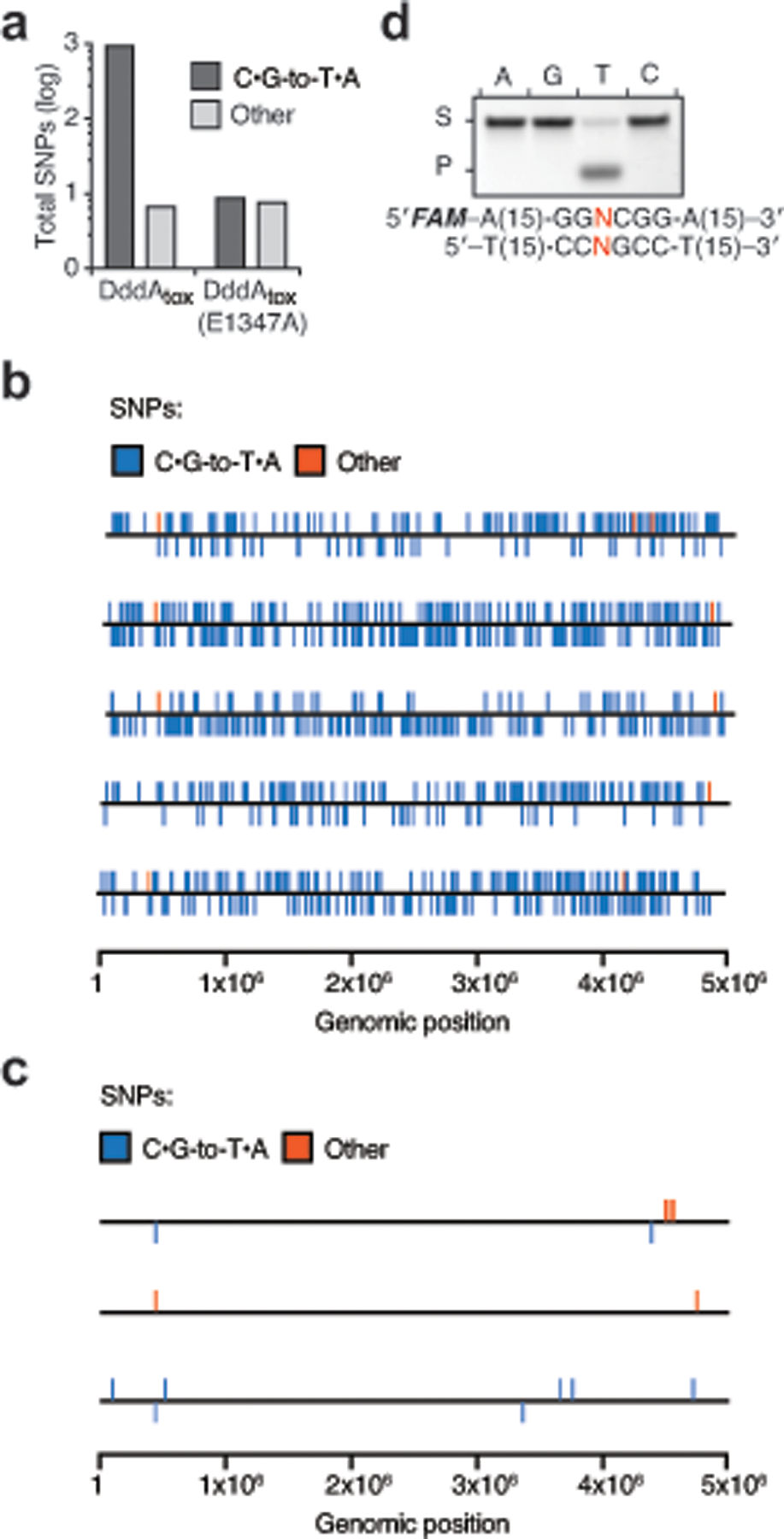
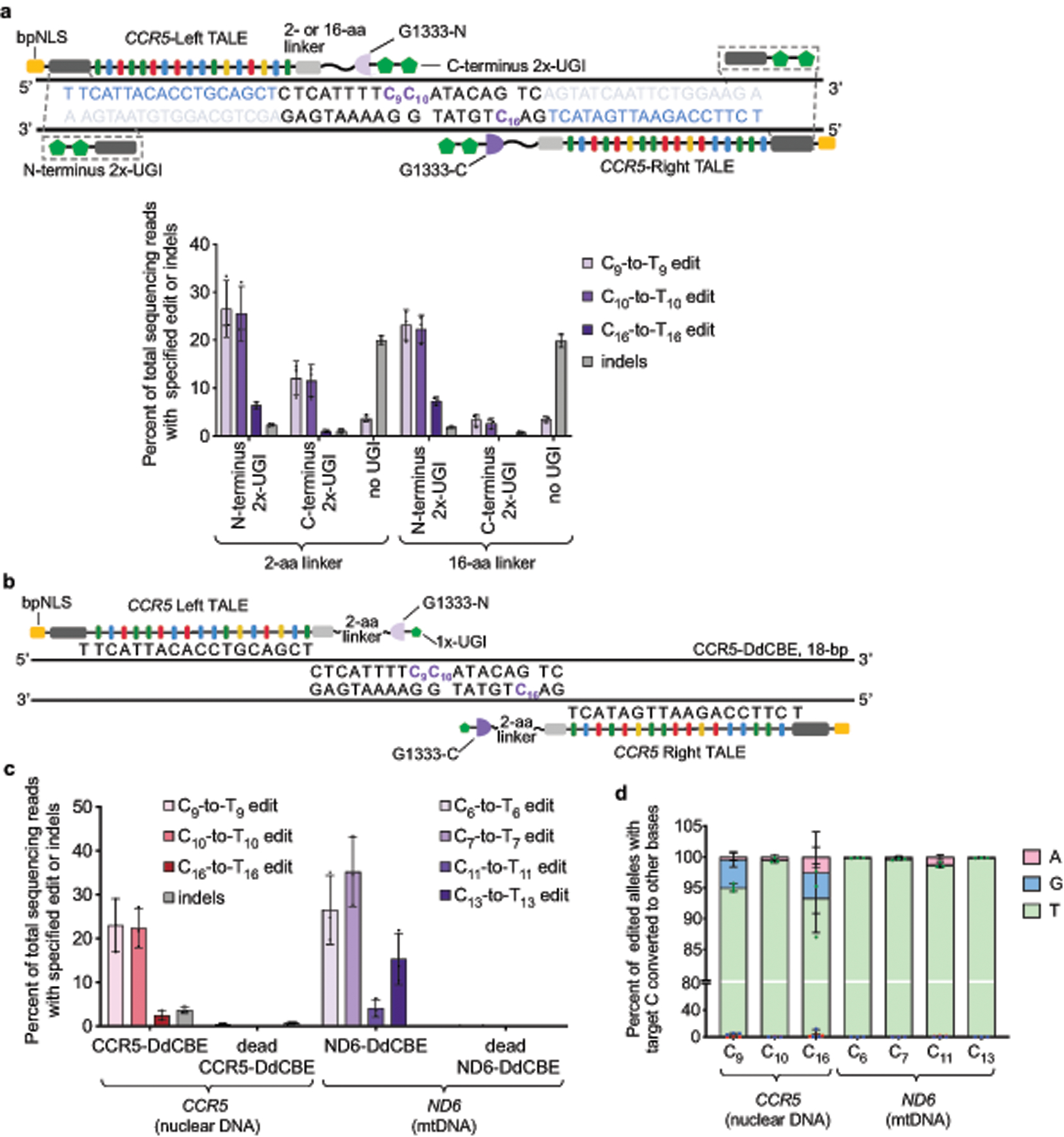
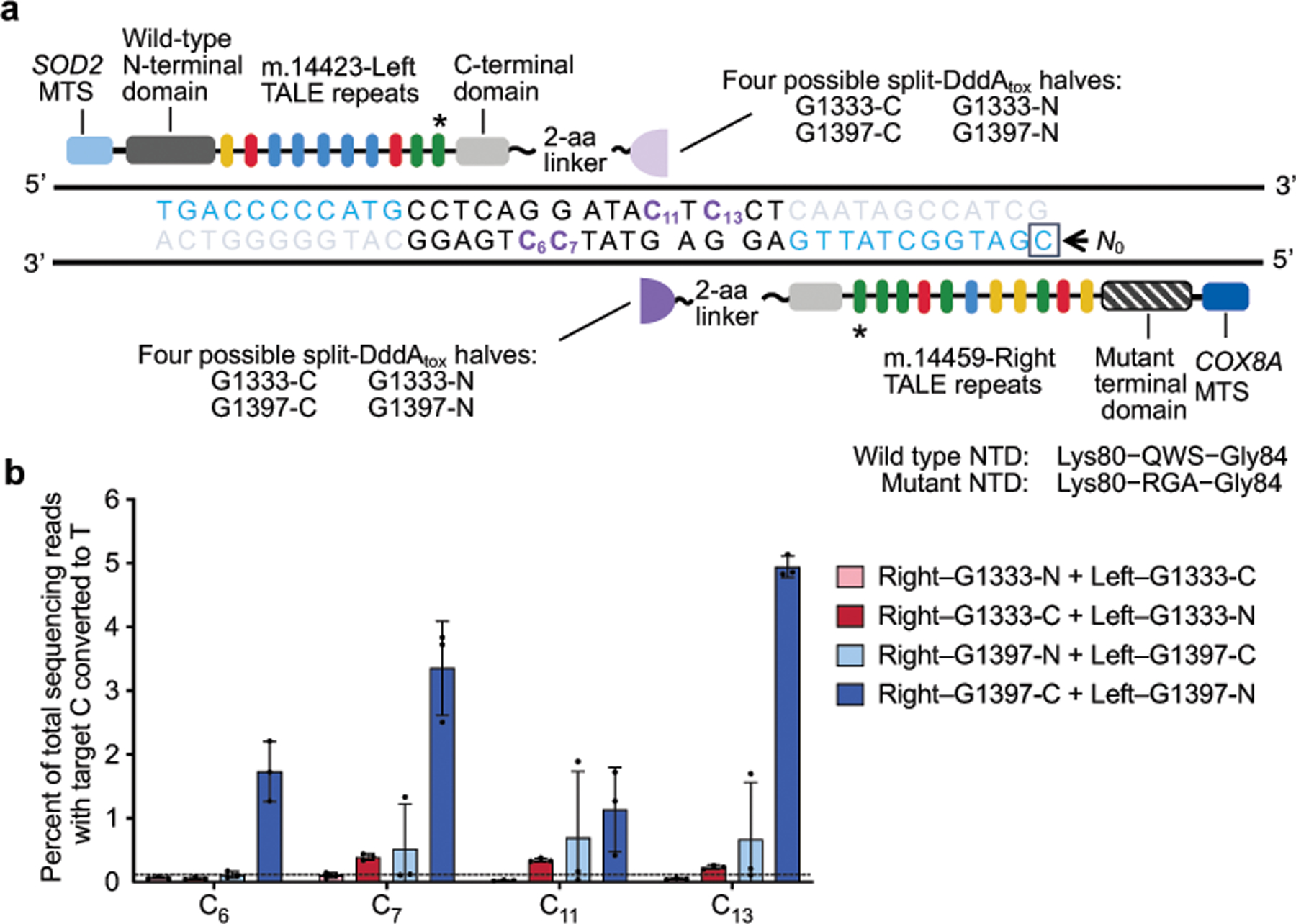
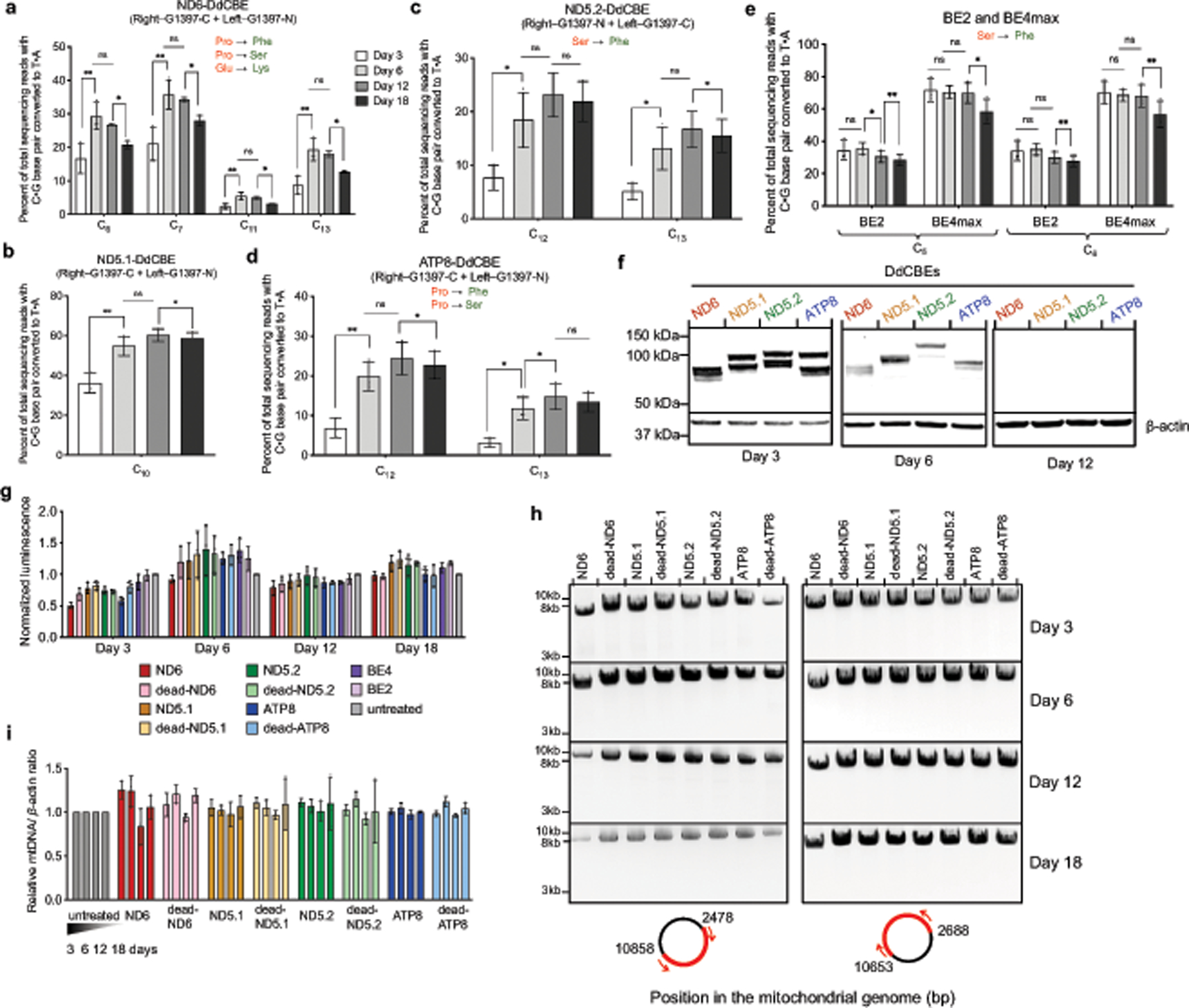
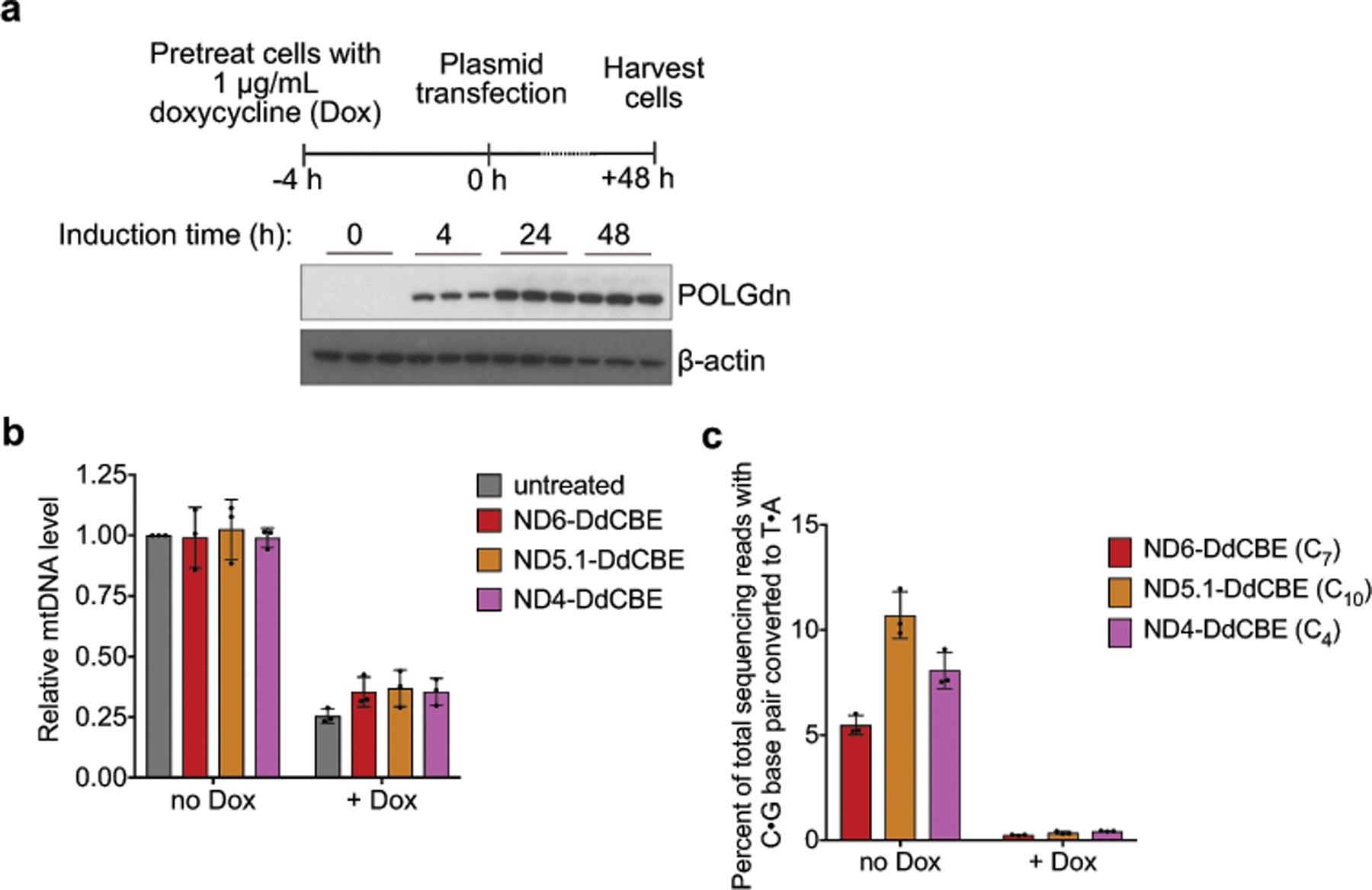
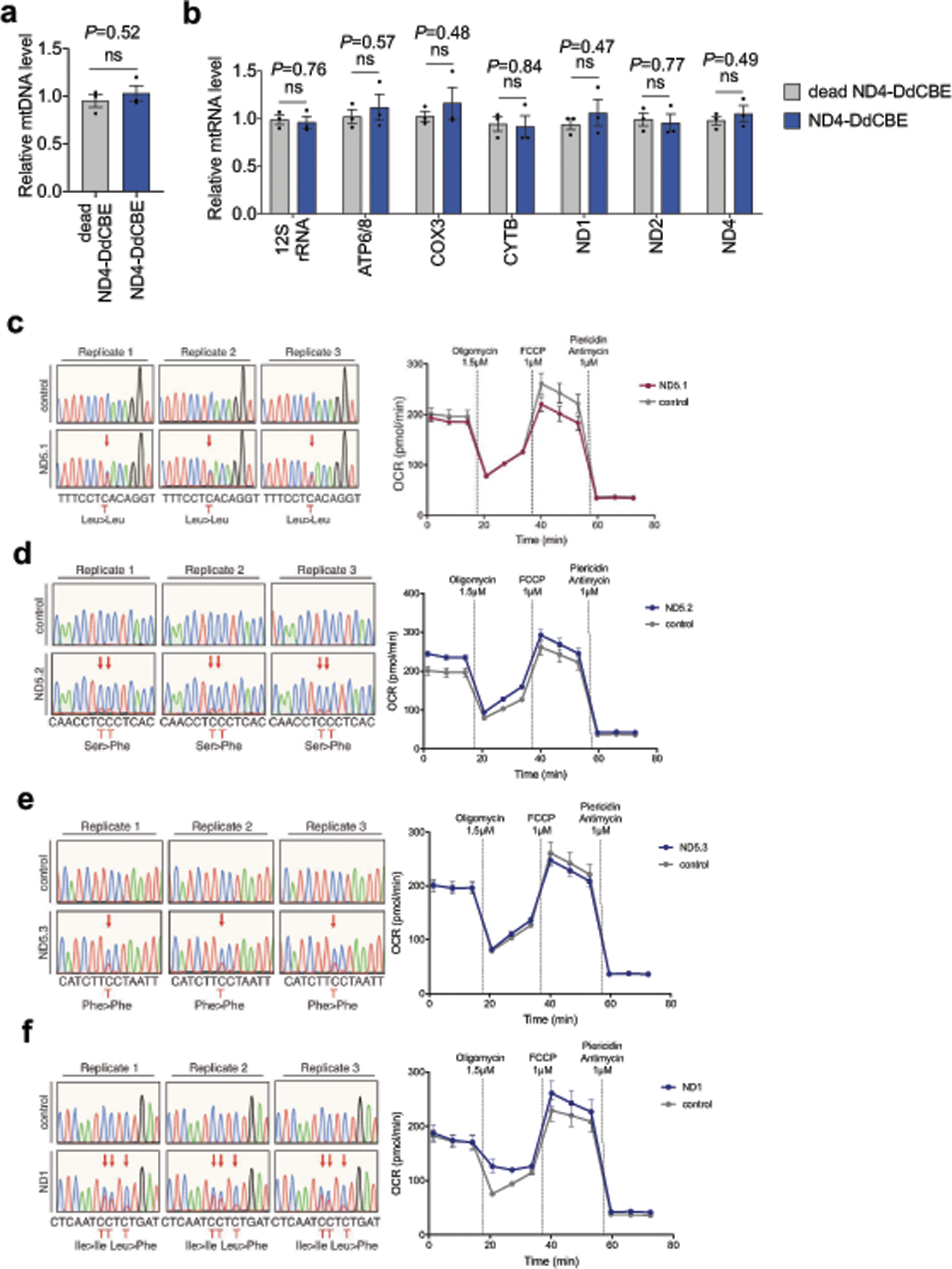
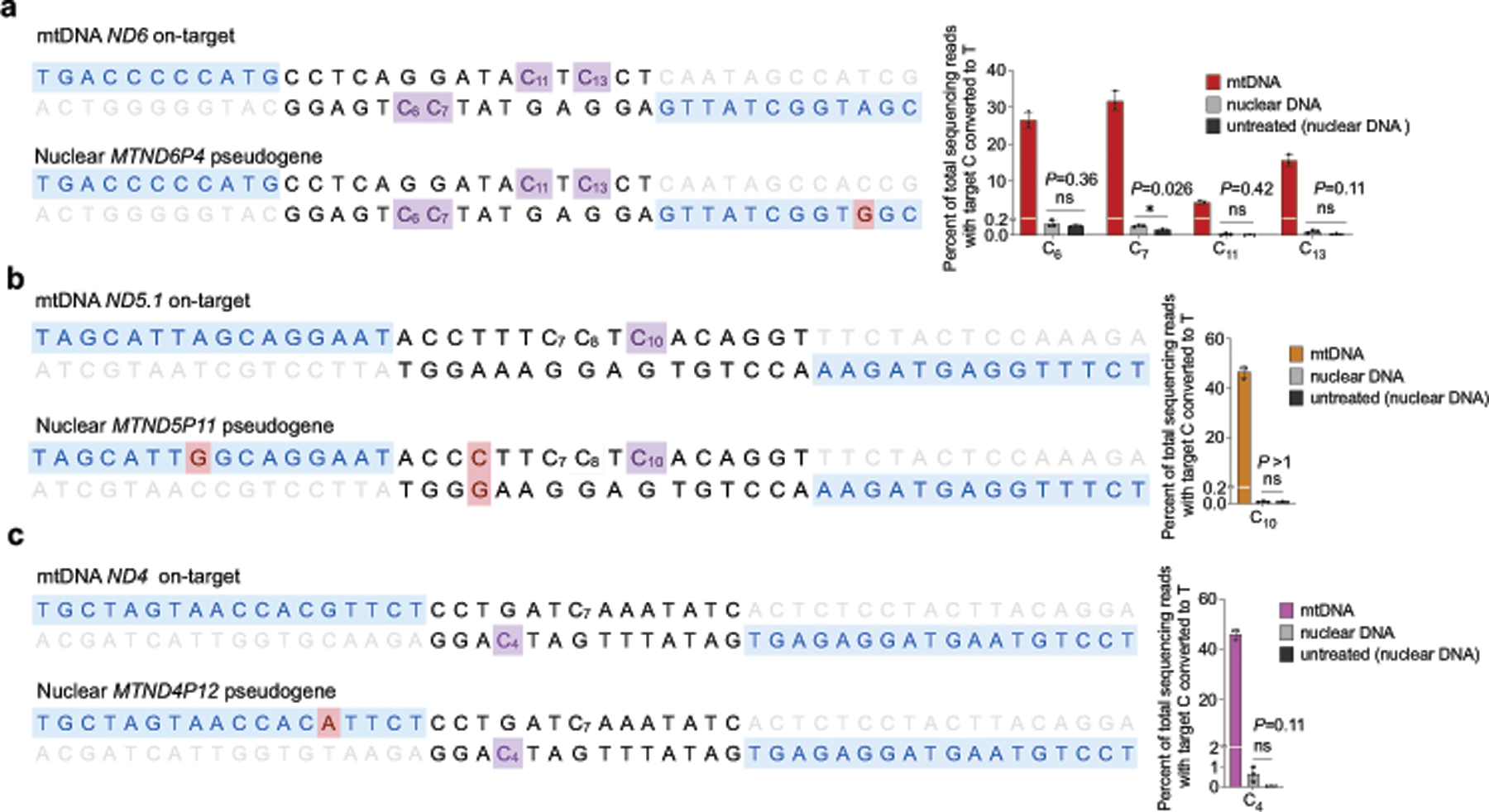
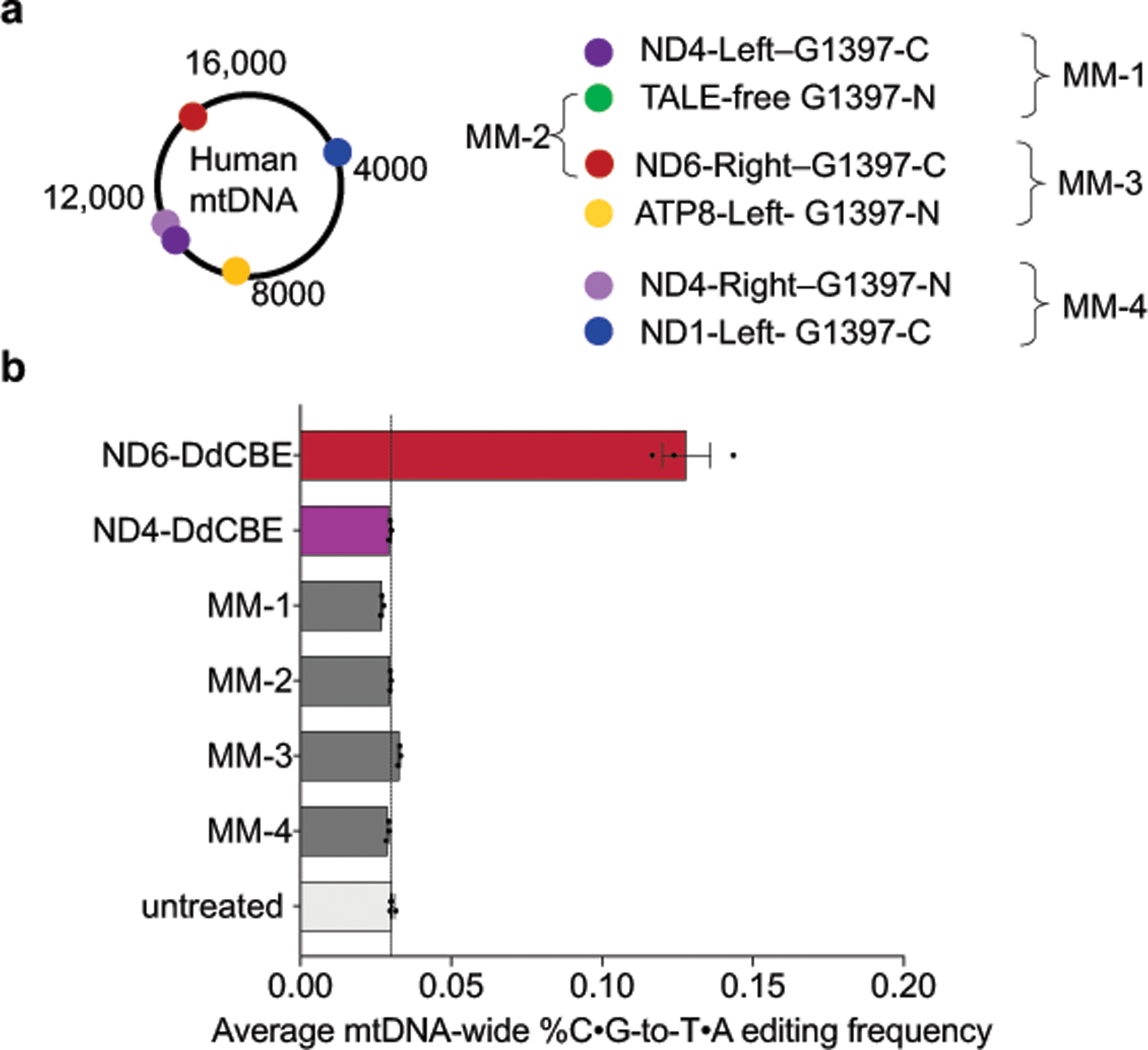
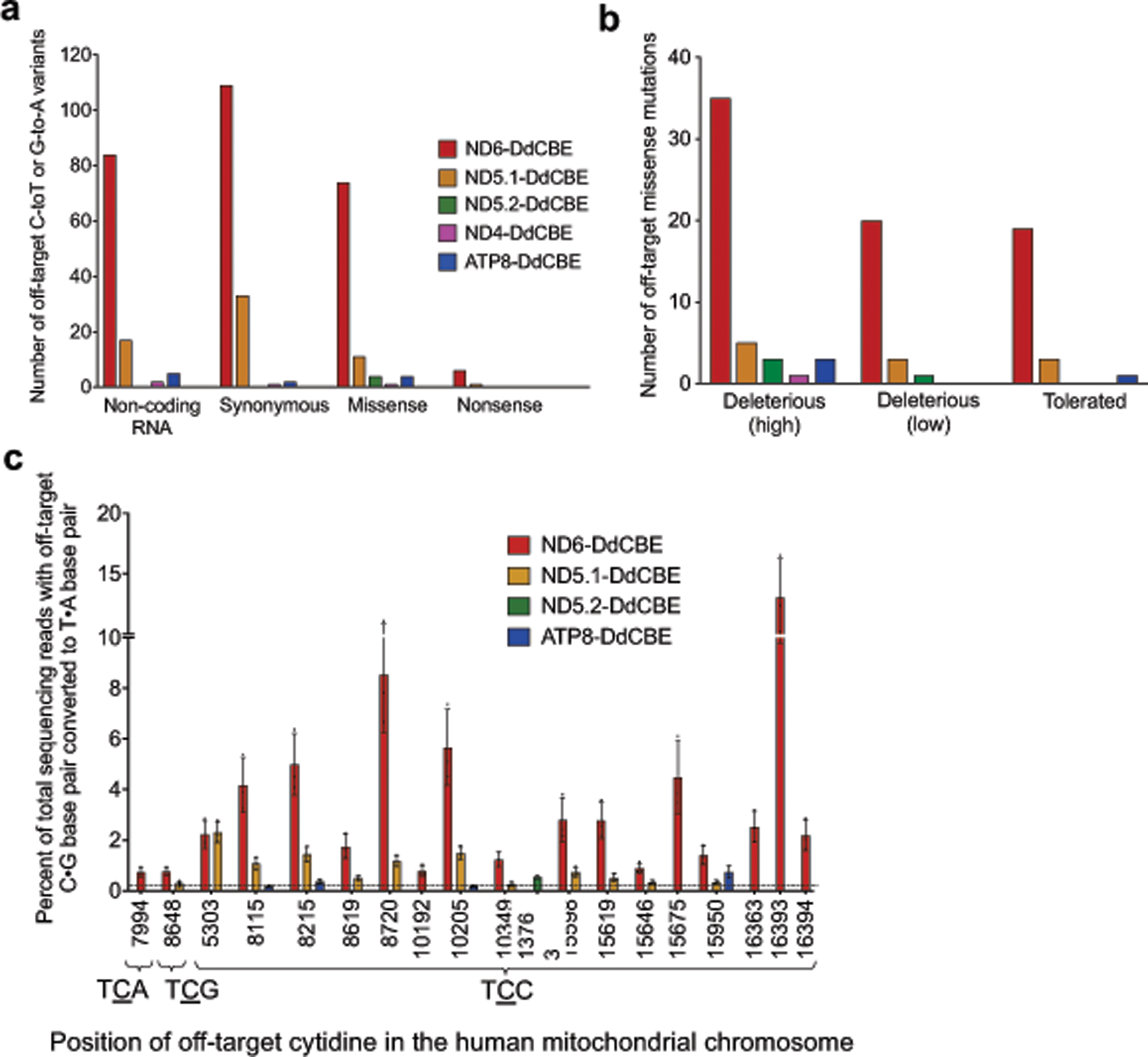
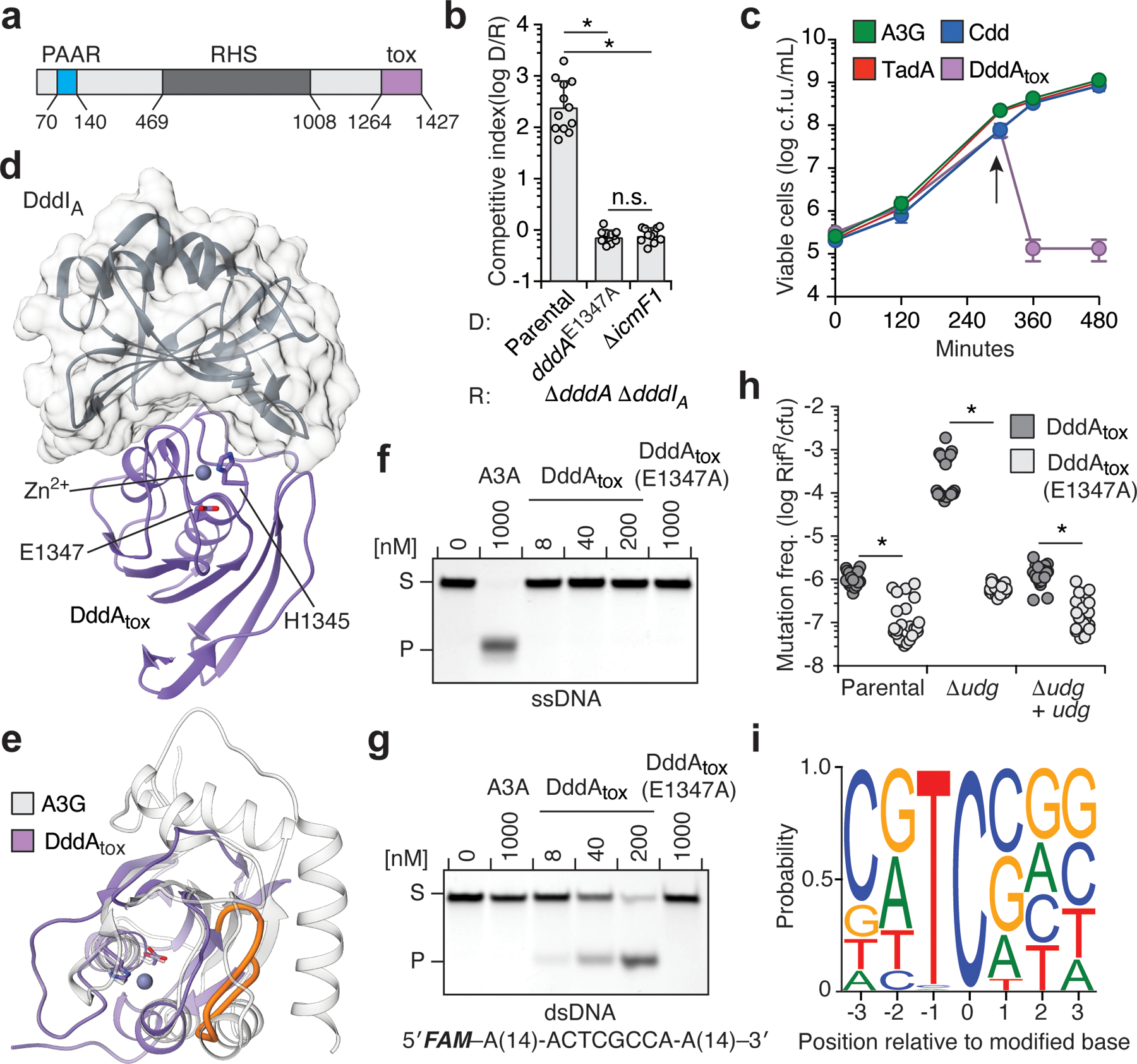
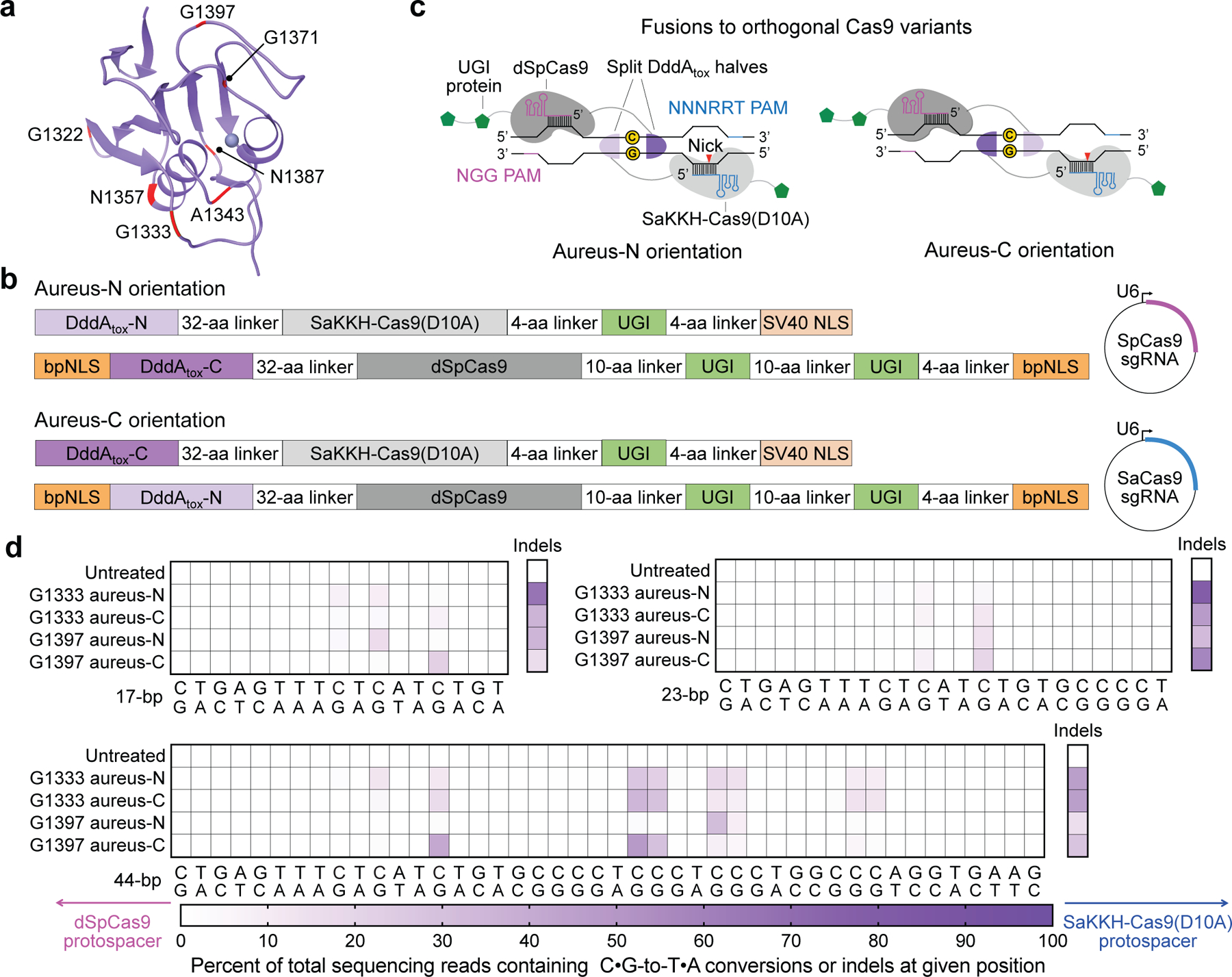
Bacterial toxins represent a vast reservoir of biochemical diversity that can be repurposed for biomedical applications. Such proteins include a group of predicted interbacterial toxins of the deaminase superfamily, members of which have found application in gene-editing techniques1,2. Because previously described cytidine deaminases operate on single-stranded nucleic acids3, their use in base editing requires the unwinding of double-stranded DNA (dsDNA)-for example by a CRISPR-Cas9 system. Base editing within mitochondrial DNA (mtDNA), however, has thus far been hindered by challenges associated with the delivery of guide RNA into the mitochondria4. As a consequence, manipulation of mtDNA to date has been limited to the targeted destruction of the mitochondrial genome by designer nucleases9,10.Here we describe an interbacterial toxin, which we name DddA, that catalyses the deamination of cytidines within dsDNA. We engineered split-DddA halves that are non-toxic and inactive until brought together on target DNA by adjacently bound programmable DNA-binding proteins. Fusions of the split-DddA halves, transcription activator-like effector array proteins, and a uracil glycosylase inhibitor resulted in RNA-free DddA-derived cytosine base editors (DdCBEs) that catalyse C•G-to-T•A conversions in human mtDNA with high target specificity and product purity. We used DdCBEs to model a disease-associated mtDNA mutation in human cells, resulting in changes in respiration rates and oxidative phosphorylation. CRISPR-free DdCBEs enable the precise manipulation of mtDNA, rather than the elimination of mtDNA copies that results from its cleavage by targeted nucleases, with broad implications for the study and potential treatment of mitochondrial disorders.
Conflict of interest statement
Competing interests
The Broad Institute and the University of Washington have filed provisional patent applications on base-editing systems described in this study, listing B.Y.M., M.H.d.M., S.B.P., J.D.M. and D.R.L. as inventors. D.R.L. is a consultant and co-founder of Prime Medicine, Beam Therapeutics, Pairwise Plants and Editas Medicine, companies that use genome editing. V.K.M. is a consultant to 5am Ventures and Janssen Pharmaceuticals.
Figures

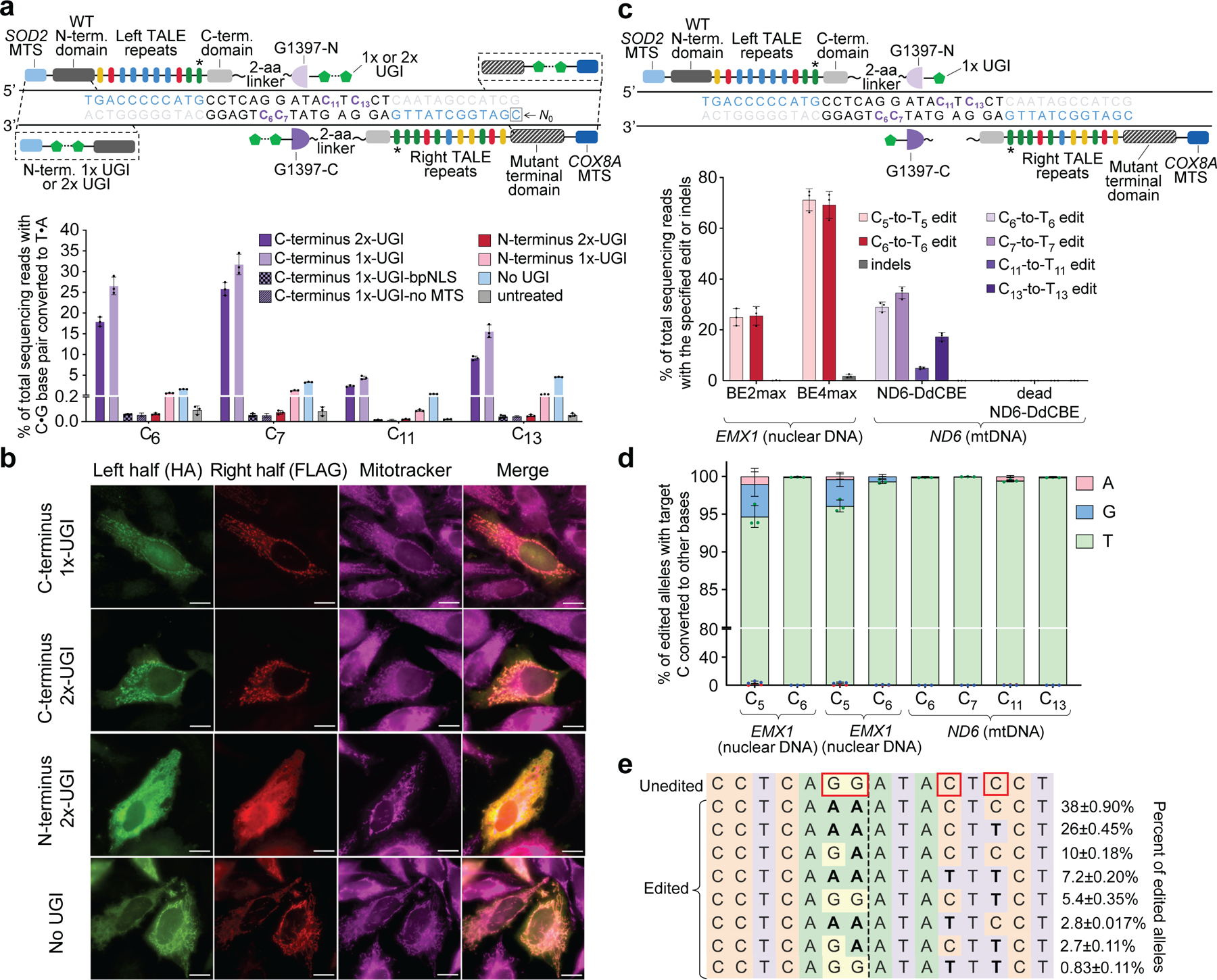
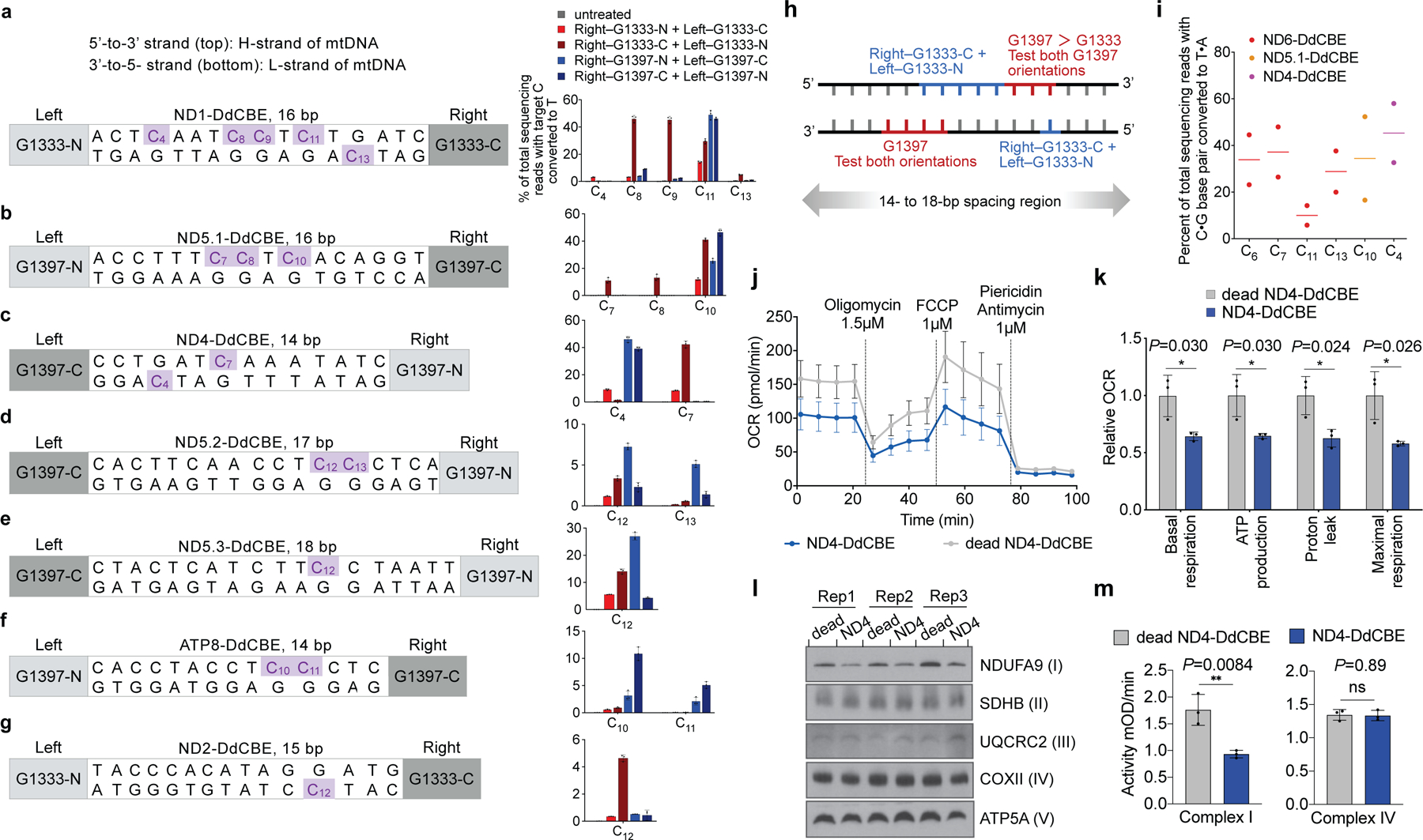
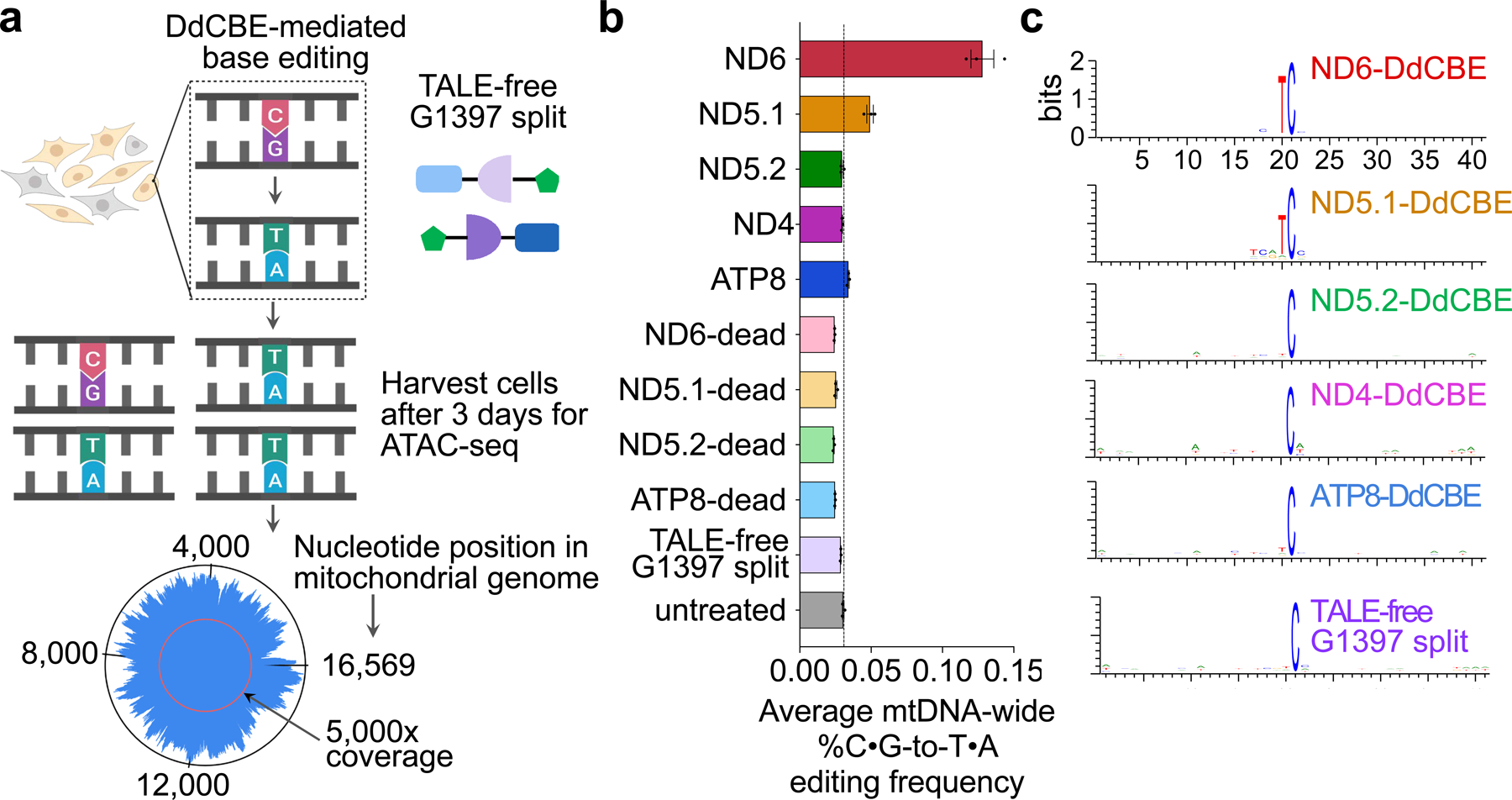
Comment in
-
Mitochondrial genome editing gets precise.Nature. 2020 Jul;583(7817):521-522. doi: 10.1038/d41586-020-01974-6. Nature. 2020. PMID: 32641789 No abstract available.
-
Scientists make precise gene edits to mitochondrial DNA for first time.Nature. 2020 Jul;583(7816):343. doi: 10.1038/d41586-020-02054-5. Nature. 2020. PMID: 32641792 No abstract available.
-
Editing the Mitochondrial Genome: No CRISPR Required.Trends Genet. 2020 Nov;36(11):809-810. doi: 10.1016/j.tig.2020.08.001. Epub 2020 Aug 17. Trends Genet. 2020. PMID: 32819722
-
Mitochondrial DNA Base Editing: Good Editing Things Still Come in Small Packages.Mol Cell. 2020 Sep 3;79(5):708-709. doi: 10.1016/j.molcel.2020.08.009. Mol Cell. 2020. PMID: 32888436
-
Editing the Mitochondrial Genome.N Engl J Med. 2020 Oct 8;383(15):1489-1491. doi: 10.1056/NEJMcibr2025332. N Engl J Med. 2020. PMID: 33027575 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
