

STXBP1 encephalopathies: Clinical spectrum, disease mechanisms, and therapeutic strategies
- PMID: 32643187
- PMCID: PMC7812771
- DOI: 10.1111/jnc.15120
STXBP1 encephalopathies: Clinical spectrum, disease mechanisms, and therapeutic strategies
Abstract
Mutations in Munc18-1/STXBP1 (syntaxin-binding protein 1) are linked to various severe early epileptic encephalopathies and neurodevelopmental disorders. Heterozygous mutations in the STXBP1 gene include missense, nonsense, frameshift, and splice site mutations, as well as intragenic deletions and duplications and whole-gene deletions. No genotype-phenotype correlation has been identified so far, and patients are treated by anti-epileptic drugs because of the lack of a specific disease-modifying therapy. The molecular disease mechanisms underlying STXBP1-linked disorders are yet to be fully understood, but both haploinsufficiency and dominant-negative mechanisms have been proposed. This review focuses on the current understanding of the phenotypic spectrum of STXBP1-linked disorders, as well as discusses disease mechanisms in the context of the numerous pathways in which STXBP1 functions in the brain. We additionally evaluate the available animal models to study these disorders and highlight potential therapeutic approaches for treating these devastating diseases.
Keywords: Munc18-1; STXBP1; encephalopathy; epilepsy; synapse; therapeutic approaches.
© 2020 International Society for Neurochemistry.
Conflict of interest statement
CONFLICTS OF INTEREST
The authors state that there was no conflict of interest in the preparation of this review.
Figures

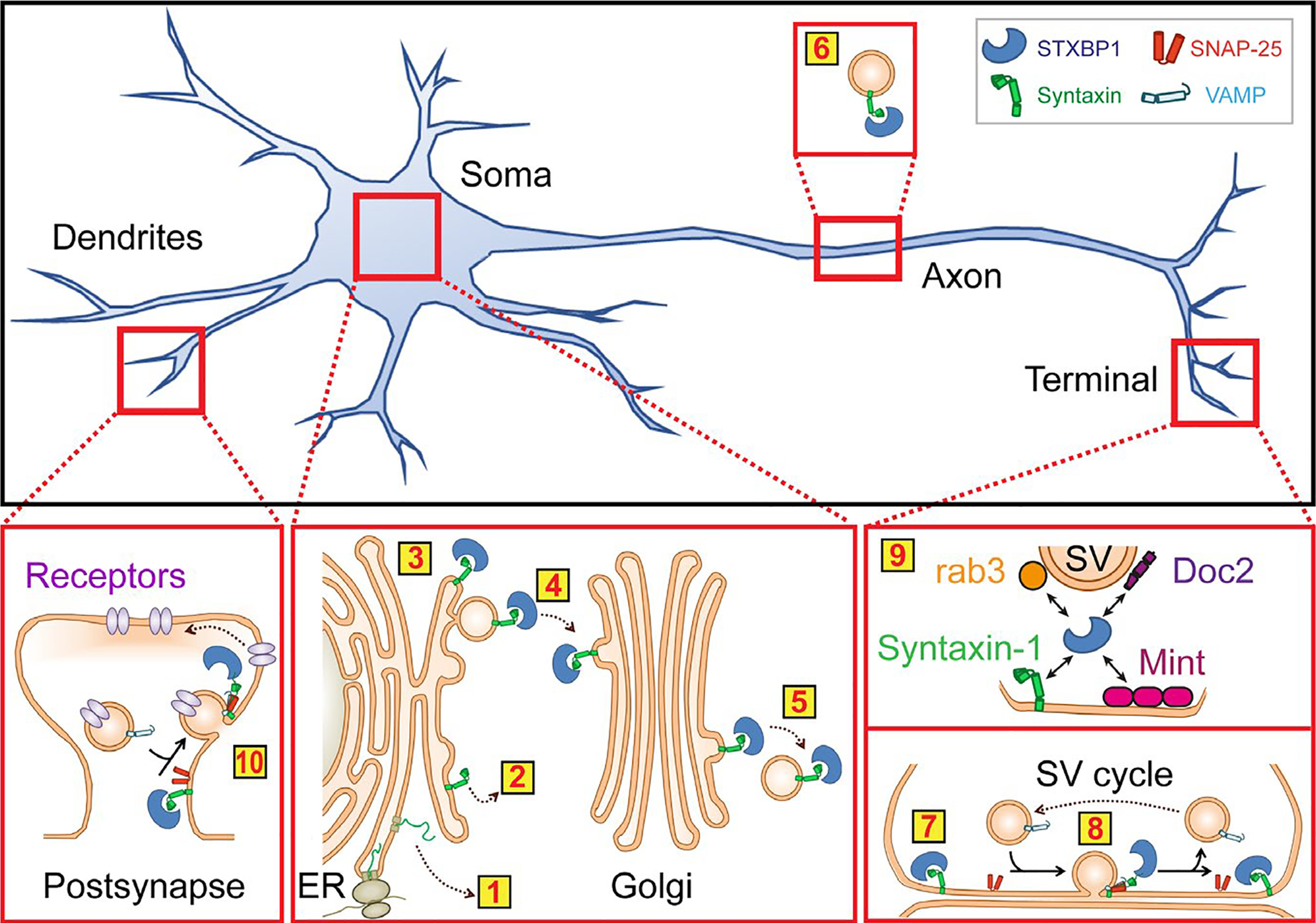

References
-
- Aalto MK, Ruohonen L, Hosono K, & Keranen S (1991). Cloning and sequencing of the yeast Saccharomyces cerevisiae SEC1 gene localized on chromosome IV. Yeast, 7, 643–650. - PubMed
-
- Alvarez Bravo G, & Yusta Izquierdo A (2018). The adult motor phenotype of Dravet syndrome is associated with mutation of the STXBP1 gene and responds well to cannabidiol treatment. Seizure, 60, 68–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
