

Analysis of tumor mutational burden: correlation of five large gene panels with whole exome sequencing
- PMID: 32647293
- PMCID: PMC7347536
- DOI: 10.1038/s41598-020-68394-4
Analysis of tumor mutational burden: correlation of five large gene panels with whole exome sequencing
Abstract
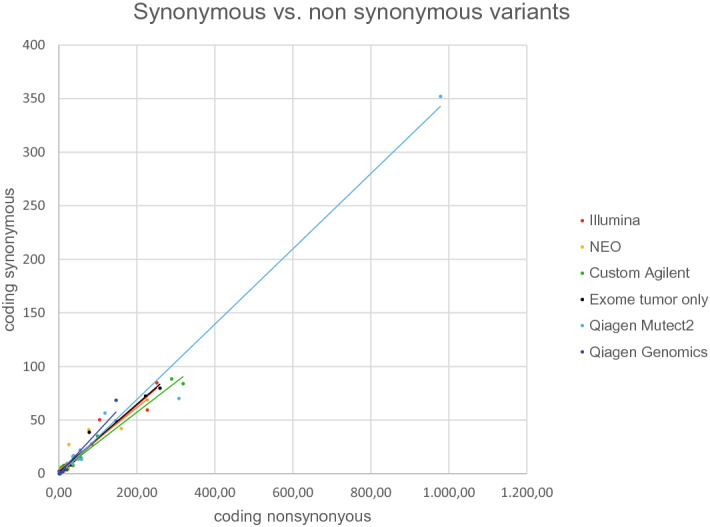
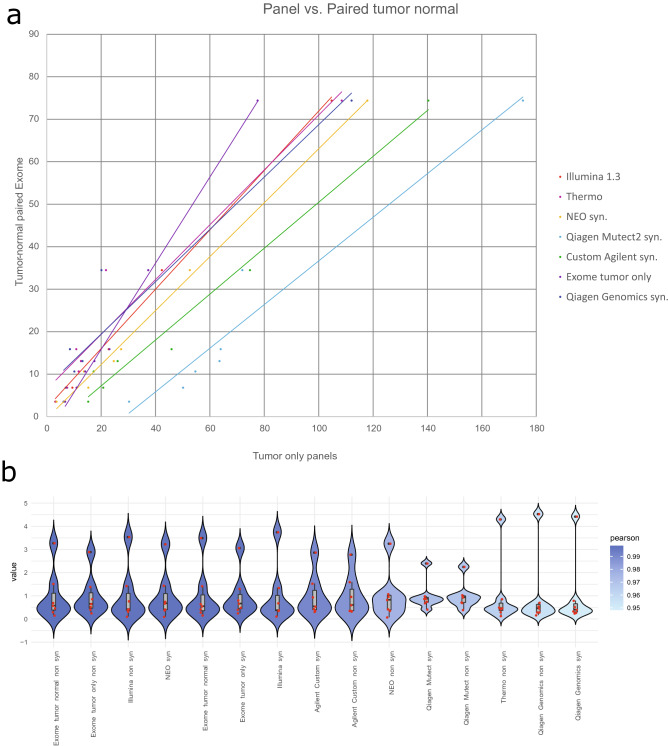
Outcome of immune checkpoint inhibition in cancer can be predicted by measuring PDL1 expression of tumor cells. Search for additional biomarkers led to tumor mutational burden (TMB) as surrogate marker for neoantigens presented. While TMB was previously determined via whole exome sequencing (WES), there have been approaches with comprehensive gene panels as well. We sequenced samples derived from formalin-fixed tumors, a POLE mutated cell line and standard DNA by WES and five different panels. If available, normal tissue was also exome sequenced. Sequencing data was analyzed by commercial software solutions and an in-house pipeline. A robust Pearson correlation (R = 0.9801 ± 0.0167; mean ± sd; N = 7) was determined for the different panels in a tumor paired normal setting for WES. Expanded analysis on tumor only exome sequenced samples yielded similar correlation (R = 0.9439 ± 0.0632; mean ± sd; N = 14). Remaining germline variants increased TMB in WES by 5.761 ± 1.953 (mean ± sd.; N = 7) variants per megabase (v/mb) for samples including synonymous variants and 3.883 ± 1.38 v/mb for samples without synonymous variants compared to tumor-normal paired calling results. Due to limited sample numbers in this study, additional replication is suggested for a clinical setting. Remaining germline variants in a tumor-only setting and artifacts caused by different library chemistries construction might affect the results.
Conflict of interest statement
The authors declare that they have no competing interests. Part of the reagents used were made available free of charge by Illumina, Qiagen and NEO New Oncology.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
