

Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma
- PMID: 32649874
- PMCID: PMC7373300
- DOI: 10.1016/j.cell.2020.06.013
Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma
Abstract
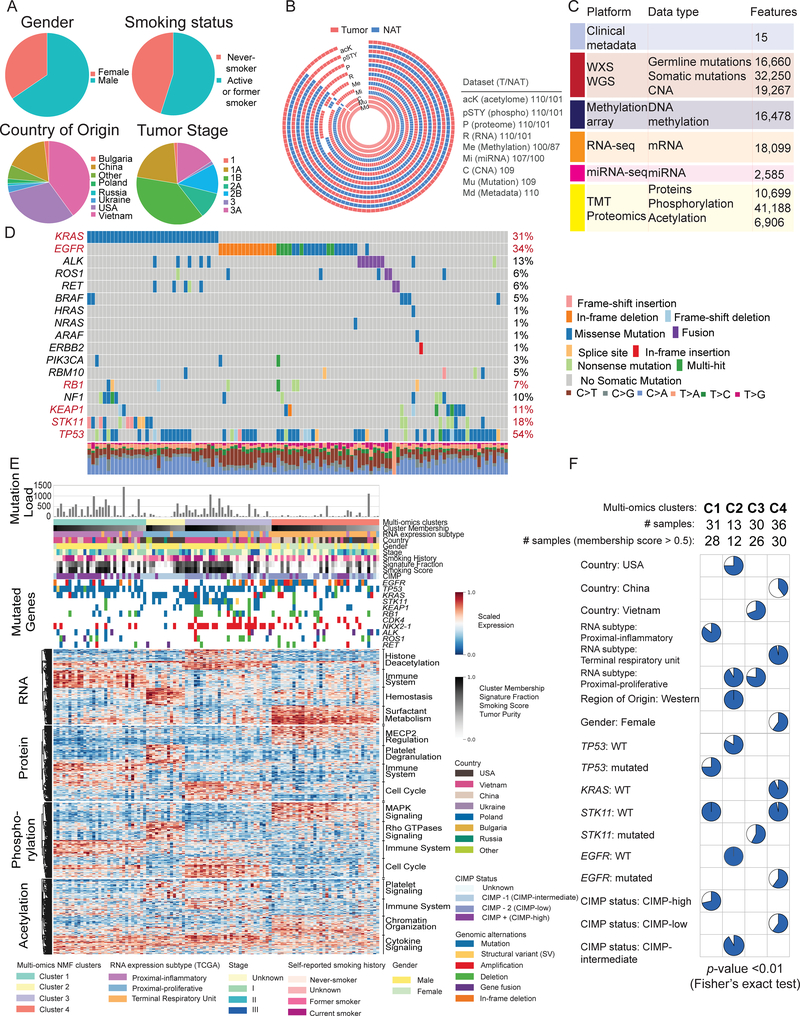
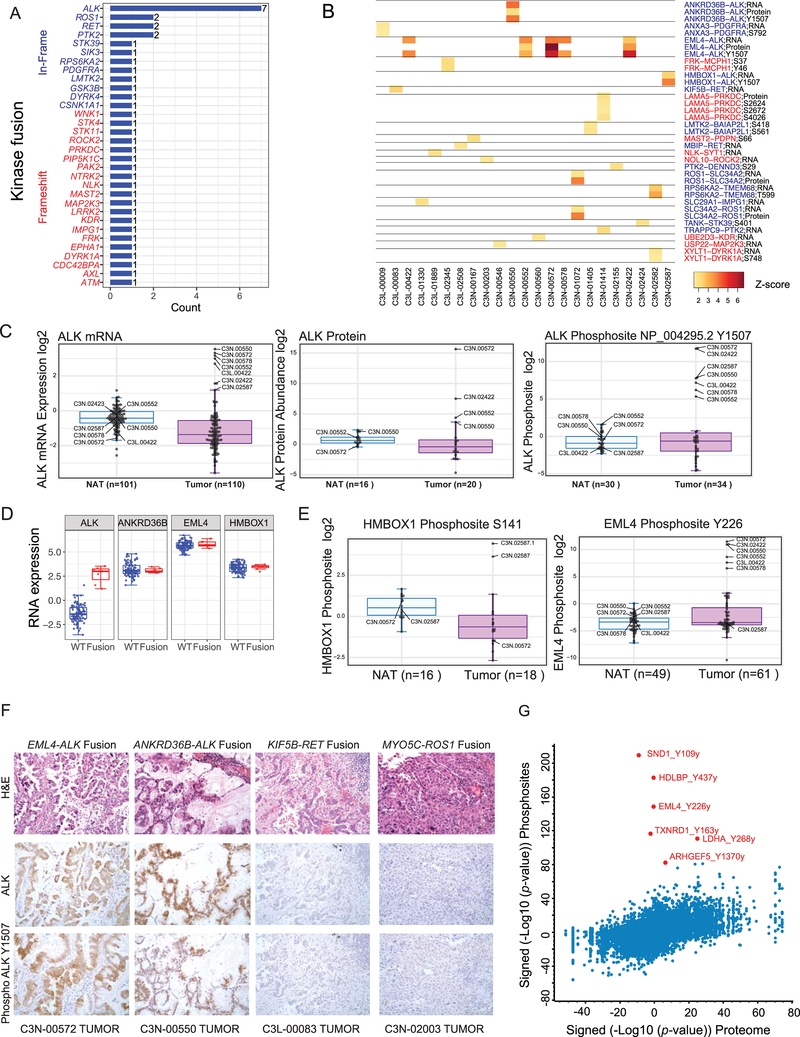
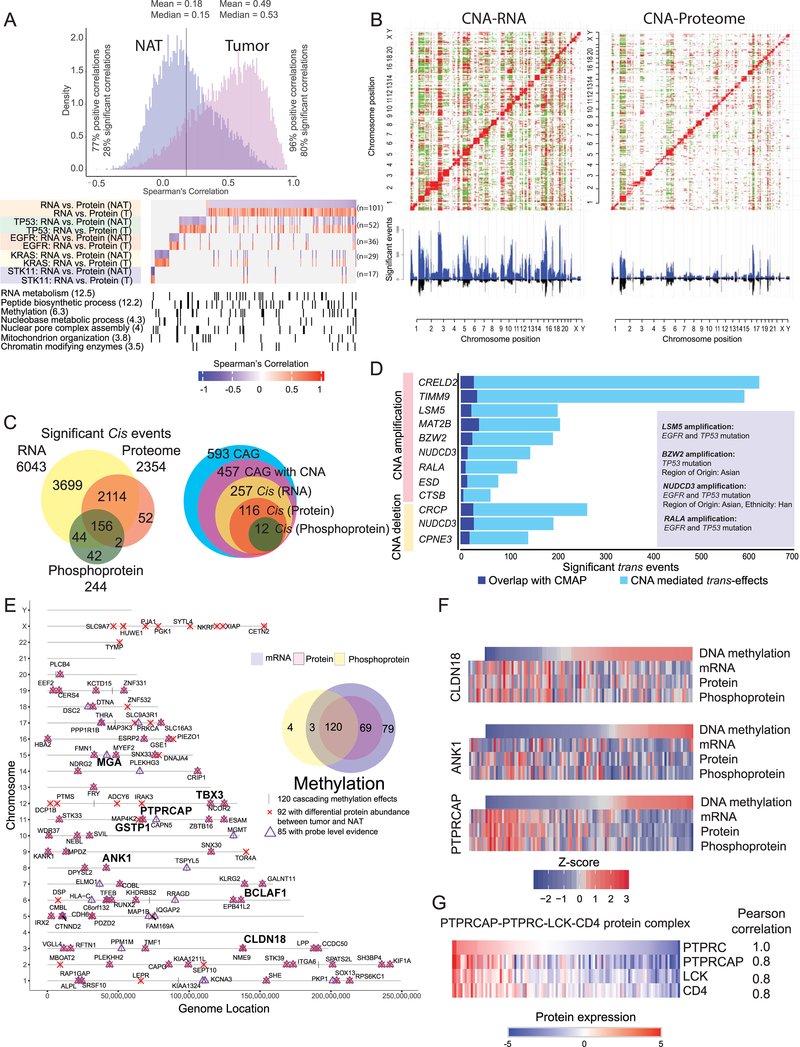
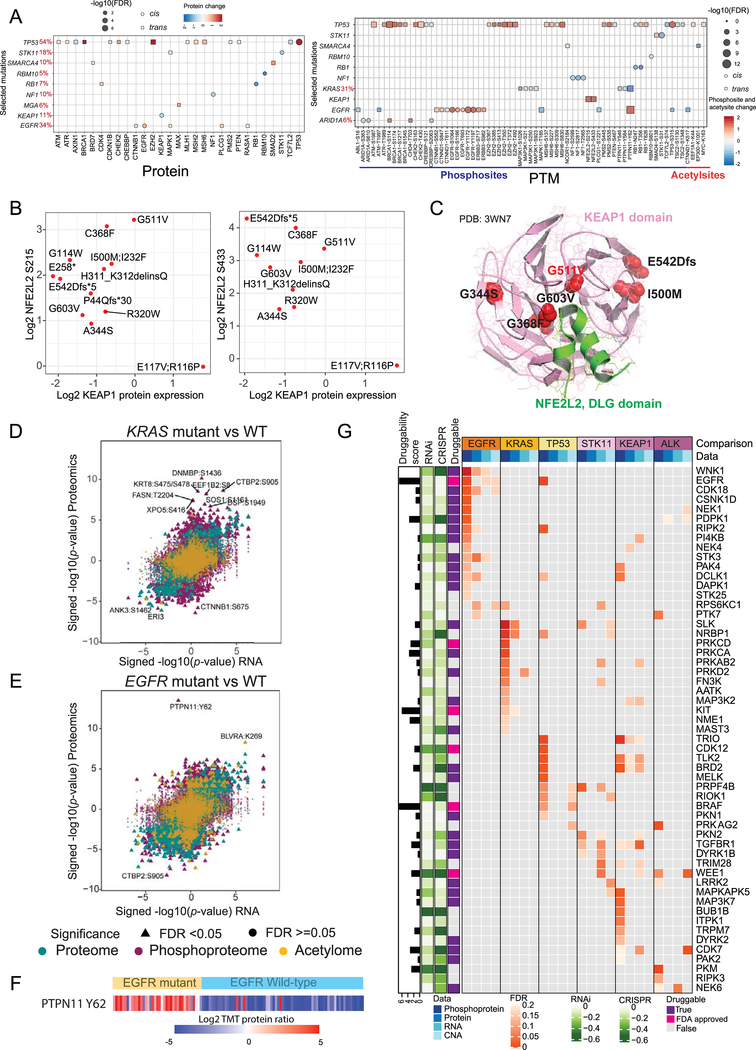
To explore the biology of lung adenocarcinoma (LUAD) and identify new therapeutic opportunities, we performed comprehensive proteogenomic characterization of 110 tumors and 101 matched normal adjacent tissues (NATs) incorporating genomics, epigenomics, deep-scale proteomics, phosphoproteomics, and acetylproteomics. Multi-omics clustering revealed four subgroups defined by key driver mutations, country, and gender. Proteomic and phosphoproteomic data illuminated biology downstream of copy number aberrations, somatic mutations, and fusions and identified therapeutic vulnerabilities associated with driver events involving KRAS, EGFR, and ALK. Immune subtyping revealed a complex landscape, reinforced the association of STK11 with immune-cold behavior, and underscored a potential immunosuppressive role of neutrophil degranulation. Smoking-associated LUADs showed correlation with other environmental exposure signatures and a field effect in NATs. Matched NATs allowed identification of differentially expressed proteins with potential diagnostic and therapeutic utility. This proteogenomics dataset represents a unique public resource for researchers and clinicians seeking to better understand and treat lung adenocarcinomas.
Keywords: CPTAC; acetylation; adenocarcinoma; genomics; lung cancer; mass spectrometry; phosphorylation; protein; proteogenomics; proteomics.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests B.Z. has received research funding from Bristol-Myers Squibb. All other authors have no conflict of interests to declare.
Figures
Comment in
-
Across the Globe: Proteogenomic Landscapes of Lung Cancer.Cell. 2020 Jul 9;182(1):9-11. doi: 10.1016/j.cell.2020.06.016. Cell. 2020. PMID: 32649881
-
Proteogenomics Clarifies Picture of Lung Cancer.Cancer Discov. 2020 Oct;10(10):OF3. doi: 10.1158/2159-8290.CD-NB2020-071. Epub 2020 Aug 5. Cancer Discov. 2020. PMID: 32759305
References
-
- Abril-Rodriguez G, and Ribas A (2017). Snapshot: Immune Checkpoint Inhibitors. Cancer Cell 31, 848–848.e1. - PubMed
-
- Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G, Confalonieri S, Quarto M, Hu G, Balwierz PJ, et al. (2012). Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat. Med 18, 529–537. - PubMed
-
- Allard D, Allard B, Gaudreau P-O, Chrobak P, and Stagg J (2016a). CD73-adenosine: a next-generation target in immuno-oncology. Immunotherapy 8, 145–163. - PubMed
-
- Allard D, Allard B, Gaudreau P-O, Chrobak P, and Stagg J (2016b). CD73-adenosine: a next-generation target in immuno-oncology. Immunotherapy 8, 145–163. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U24 CA210954/CA/NCI NIH HHS/United States
- P30 ES010126/ES/NIEHS NIH HHS/United States
- U24 CA210985/CA/NCI NIH HHS/United States
- U24 CA210967/CA/NCI NIH HHS/United States
- U24 CA210986/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- U24 CA210972/CA/NCI NIH HHS/United States
- P30 ES017885/ES/NIEHS NIH HHS/United States
- U24 CA210993/CA/NCI NIH HHS/United States
- U24 CA210979/CA/NCI NIH HHS/United States
- U01 CA214125/CA/NCI NIH HHS/United States
- U24 CA210955/CA/NCI NIH HHS/United States
- P20 GM130423/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
