

Oxidative Stress in Cancer
- PMID: 32649885
- PMCID: PMC7439808
- DOI: 10.1016/j.ccell.2020.06.001
Oxidative Stress in Cancer
Abstract
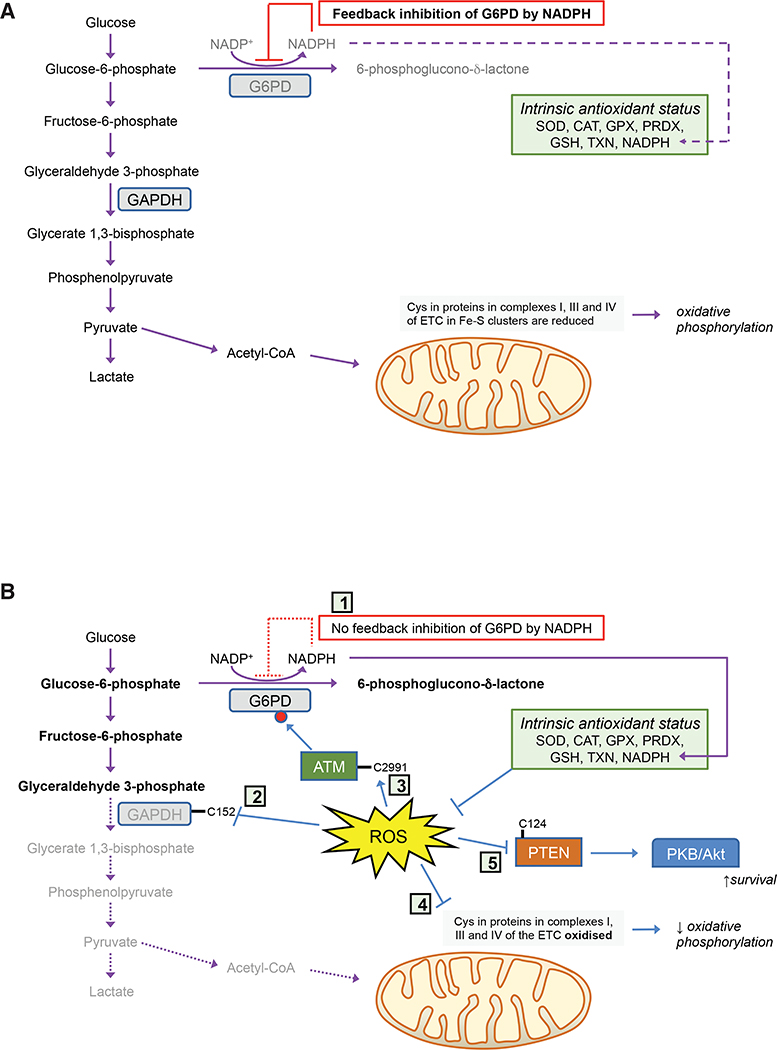
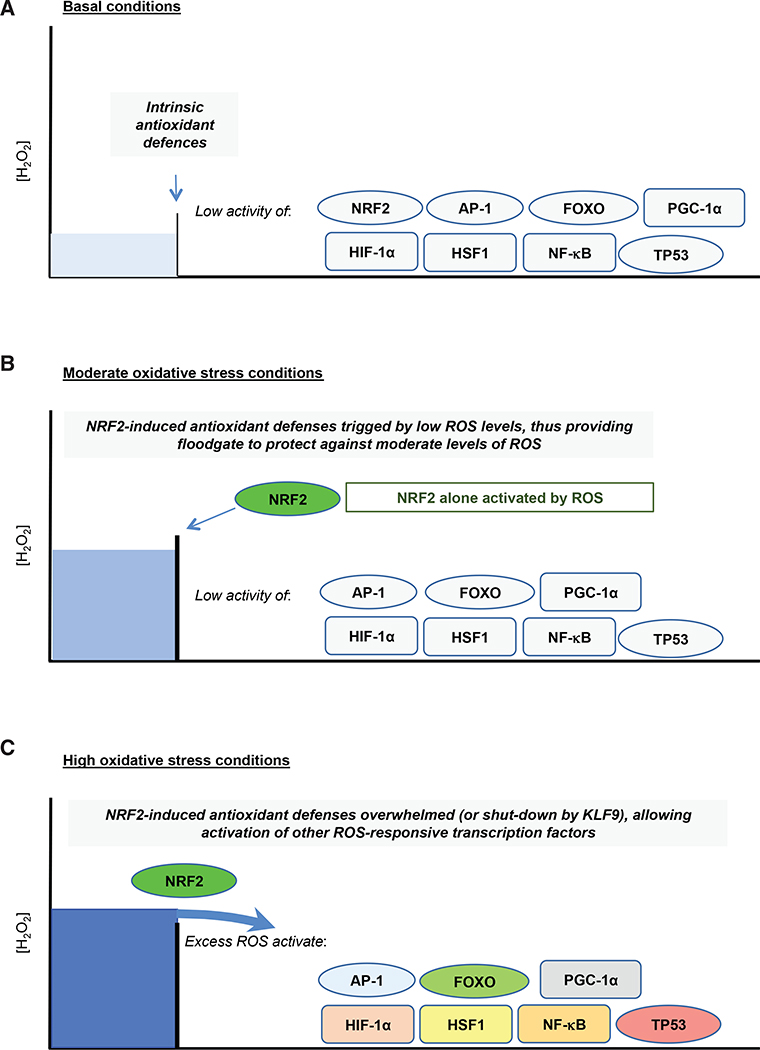
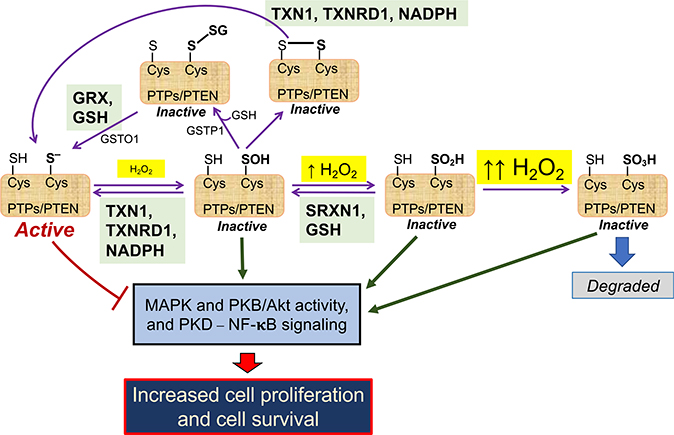
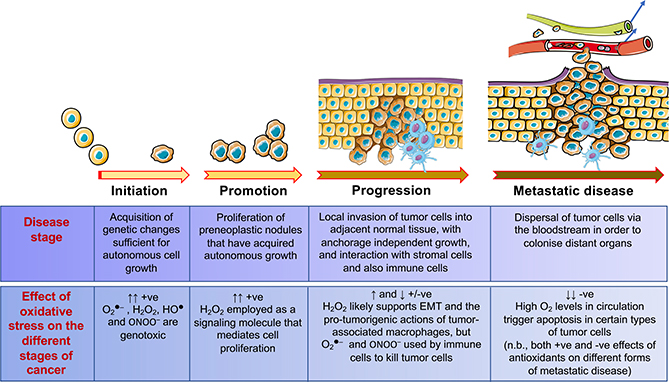
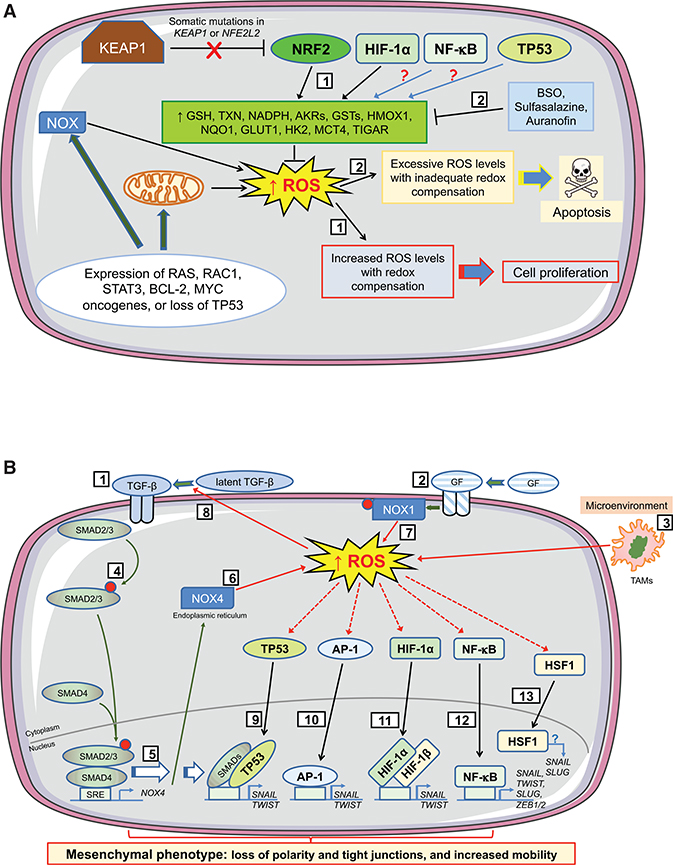
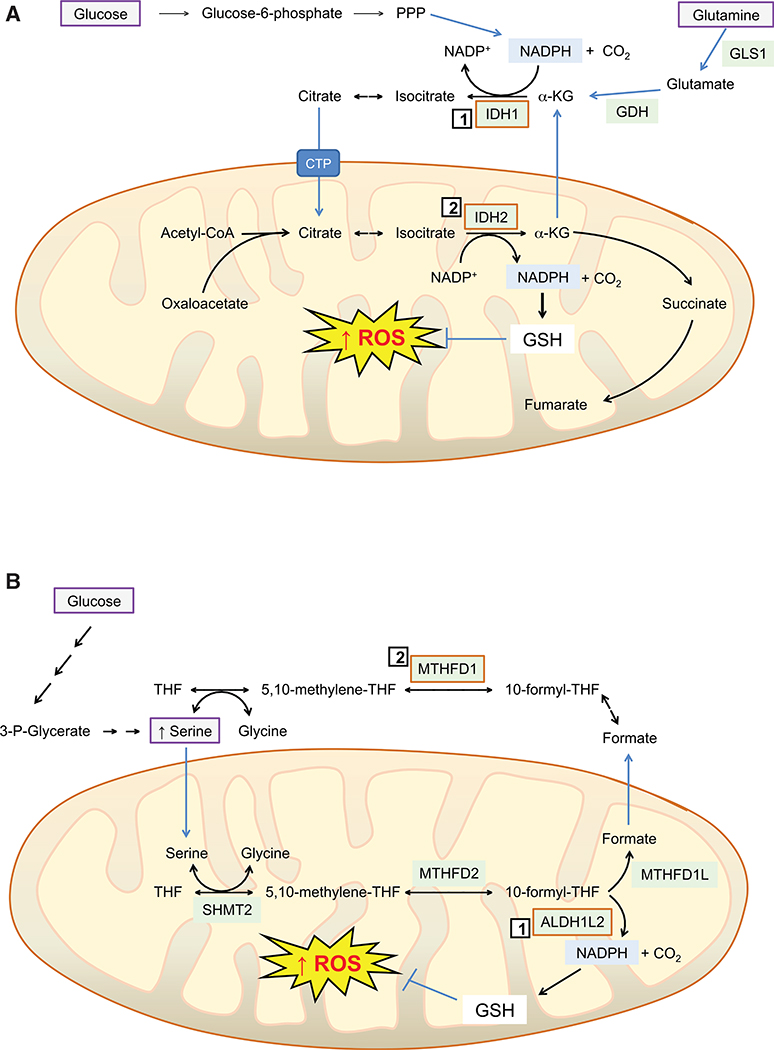
Contingent upon concentration, reactive oxygen species (ROS) influence cancer evolution in apparently contradictory ways, either initiating/stimulating tumorigenesis and supporting transformation/proliferation of cancer cells or causing cell death. To accommodate high ROS levels, tumor cells modify sulfur-based metabolism, NADPH generation, and the activity of antioxidant transcription factors. During initiation, genetic changes enable cell survival under high ROS levels by activating antioxidant transcription factors or increasing NADPH via the pentose phosphate pathway (PPP). During progression and metastasis, tumor cells adapt to oxidative stress by increasing NADPH in various ways, including activation of AMPK, the PPP, and reductive glutamine and folate metabolism.
Keywords: AP-1; BACH1; FOXO; HIF-1alpha; HSF1; NADPH generation; NF-κB; NRF2; PGC-1alpha; TP53; adaptation; antioxidant; dormant cancer cell; folate metabolism; glutathione; initiation; metastasis; oxidative stress; pentose phosphate pathway; progression; reactive oxygen species; recurrent disease; redox signaling; reductive glutamine metabolism; thioredoxin; tumorigenesis.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests A.D.K. is on the Scientific Advisory Board of Evgen Pharma and is a consultant for Aclipse Therapeutics and Vividion Therapeutics.
Figures
References
-
- Alexander MS, Wilkes JG, Schroeder SR, Buettner GR, Wagner BA, Du J, Gibson-Corley K, O’Leary BR, Spitz DR, Buatti JM, et al. (2018). Pharmacologic ascorbate reduces radiation-induced normal tissue toxicity and enhances tumor radiosensitization in pancreatic cancer. Cancer Res. 78, 6838–6851. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
