

Parallel Chemoselective Profiling for Mapping Protein Structure
- PMID: 32649906
- PMCID: PMC7484201
- DOI: 10.1016/j.chembiol.2020.06.014
Parallel Chemoselective Profiling for Mapping Protein Structure
Abstract
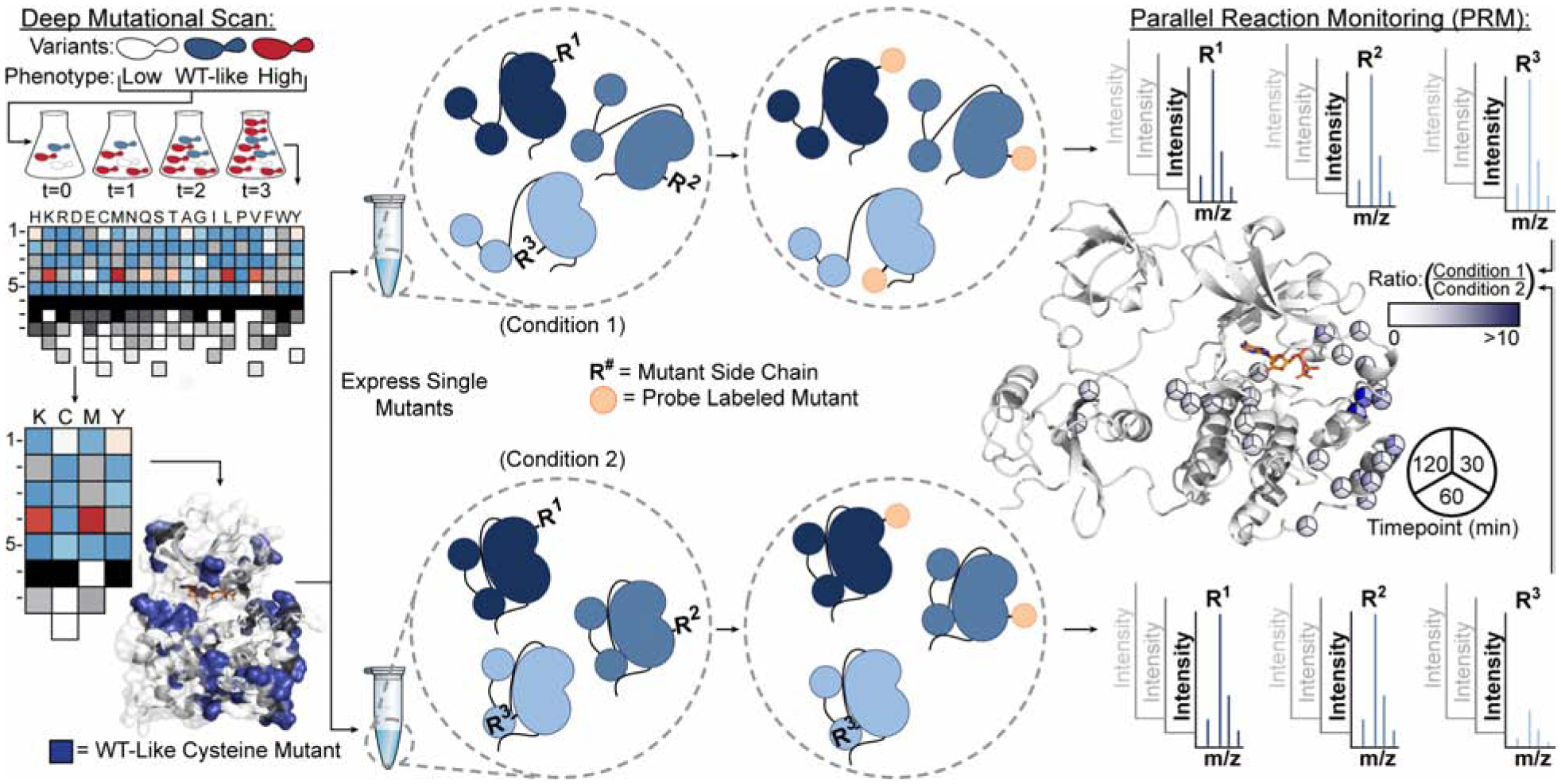
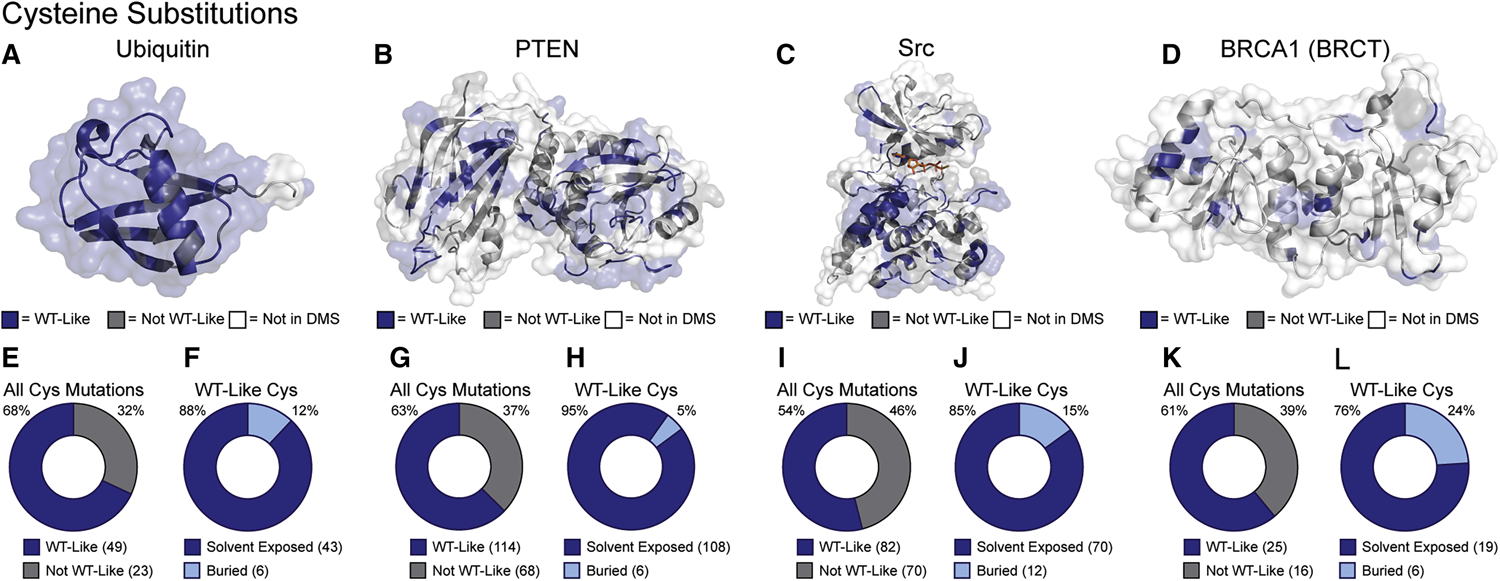
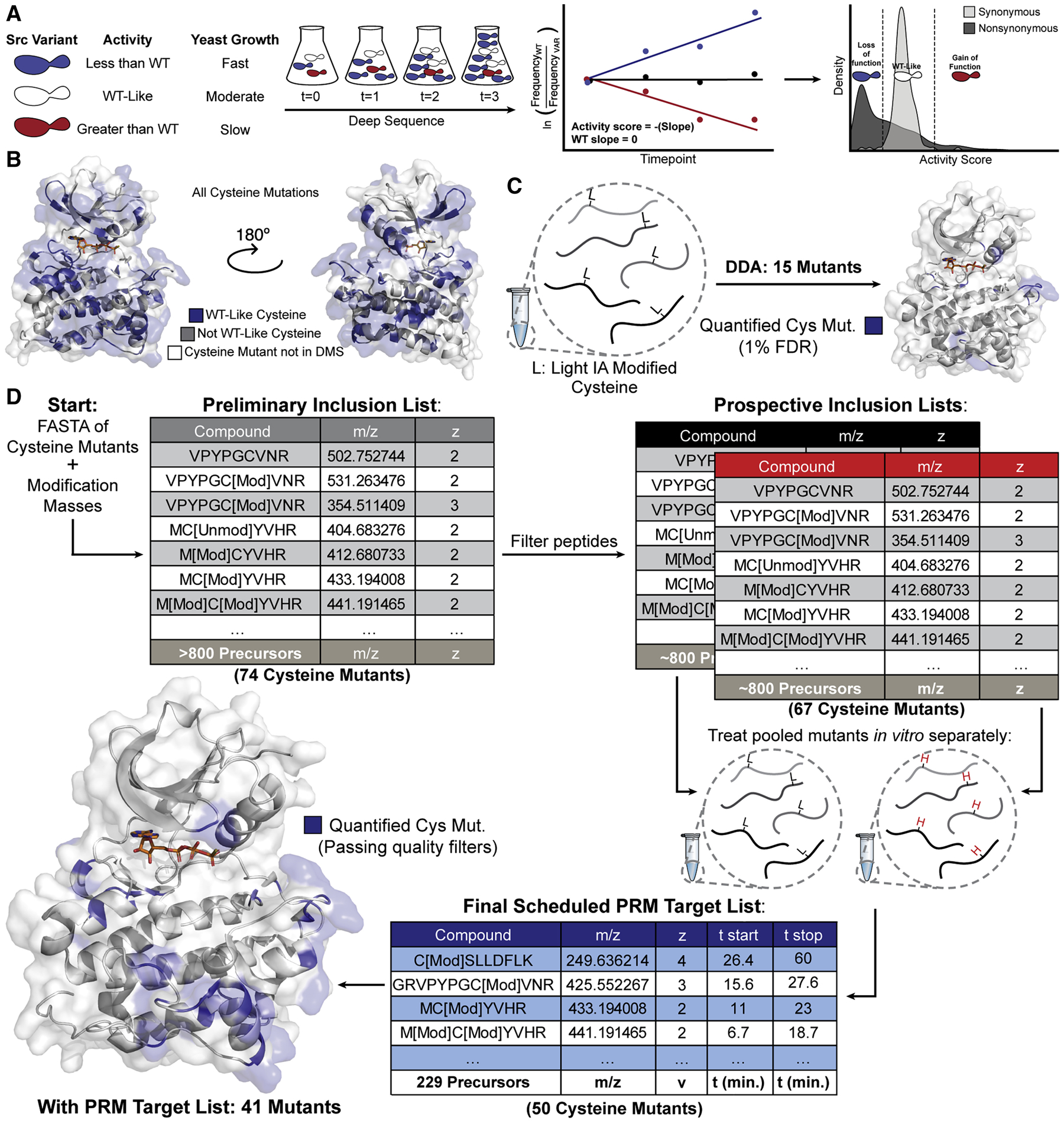
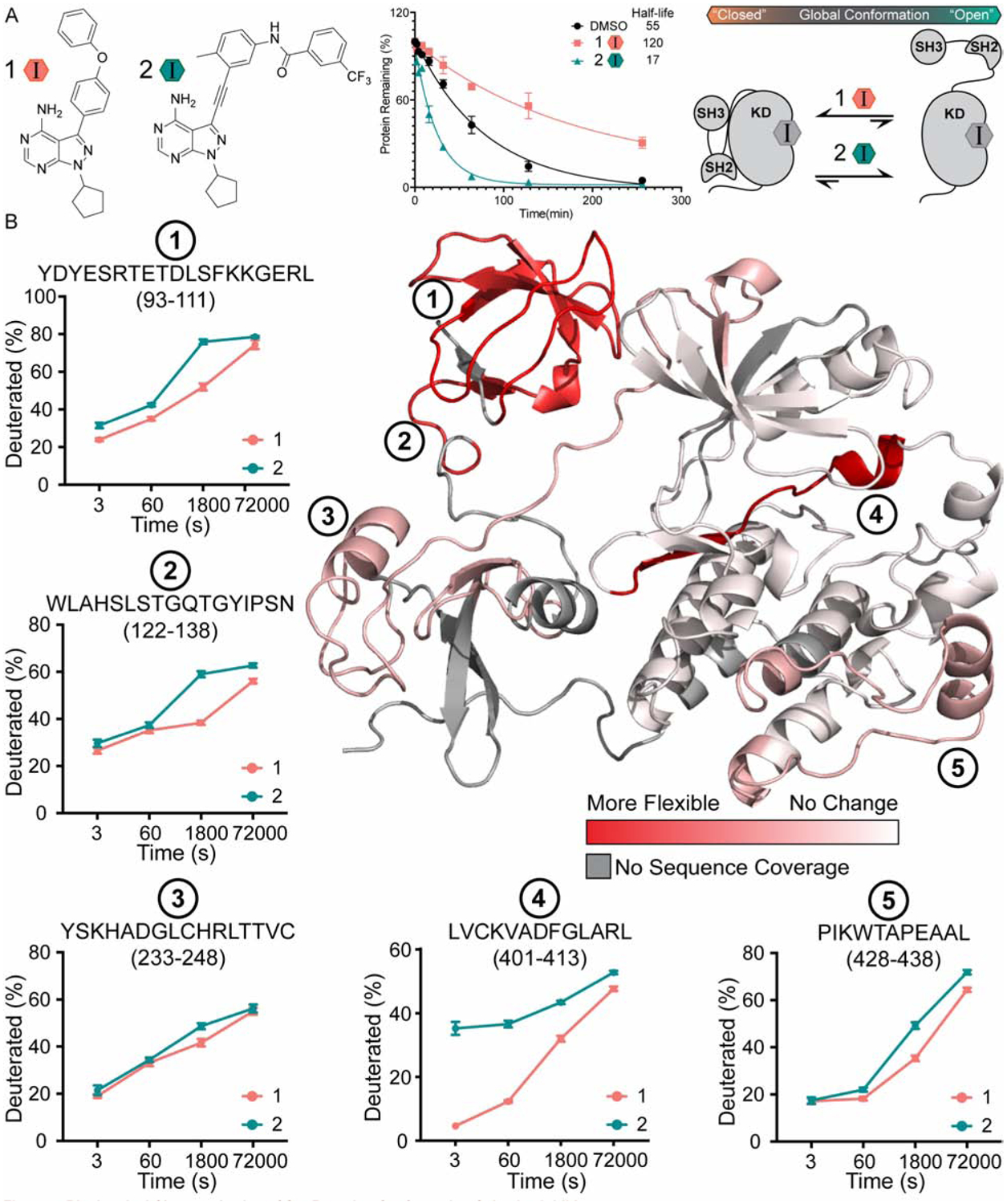
Solution-based structural techniques complement high-resolution structural data by providing insight into the oft-missed links between protein structure and dynamics. Here, we present Parallel Chemoselective Profiling, a solution-based structural method for characterizing protein structure and dynamics. Our method utilizes deep mutational scanning saturation mutagenesis data to install amino acid residues with specific chemistries at defined positions on the solvent-exposed surface of a protein. Differences in the extent of labeling of installed mutant residues are quantified using targeted mass spectrometry, reporting on each residue's local environment and structural dynamics. Using our method, we studied how conformation-selective, ATP-competitive inhibitors affect the local and global structure and dynamics of full-length Src kinase. Our results highlight how parallel chemoselective profiling can be used to study a dynamic multi-domain protein, and suggest that our method will be a useful addition to the relatively small toolkit of existing protein footprinting techniques.
Keywords: mass spectrometry; molecular dynamics; parallel chemoselective profiling; protein structure; structural proteomics.
Copyright © 2020 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
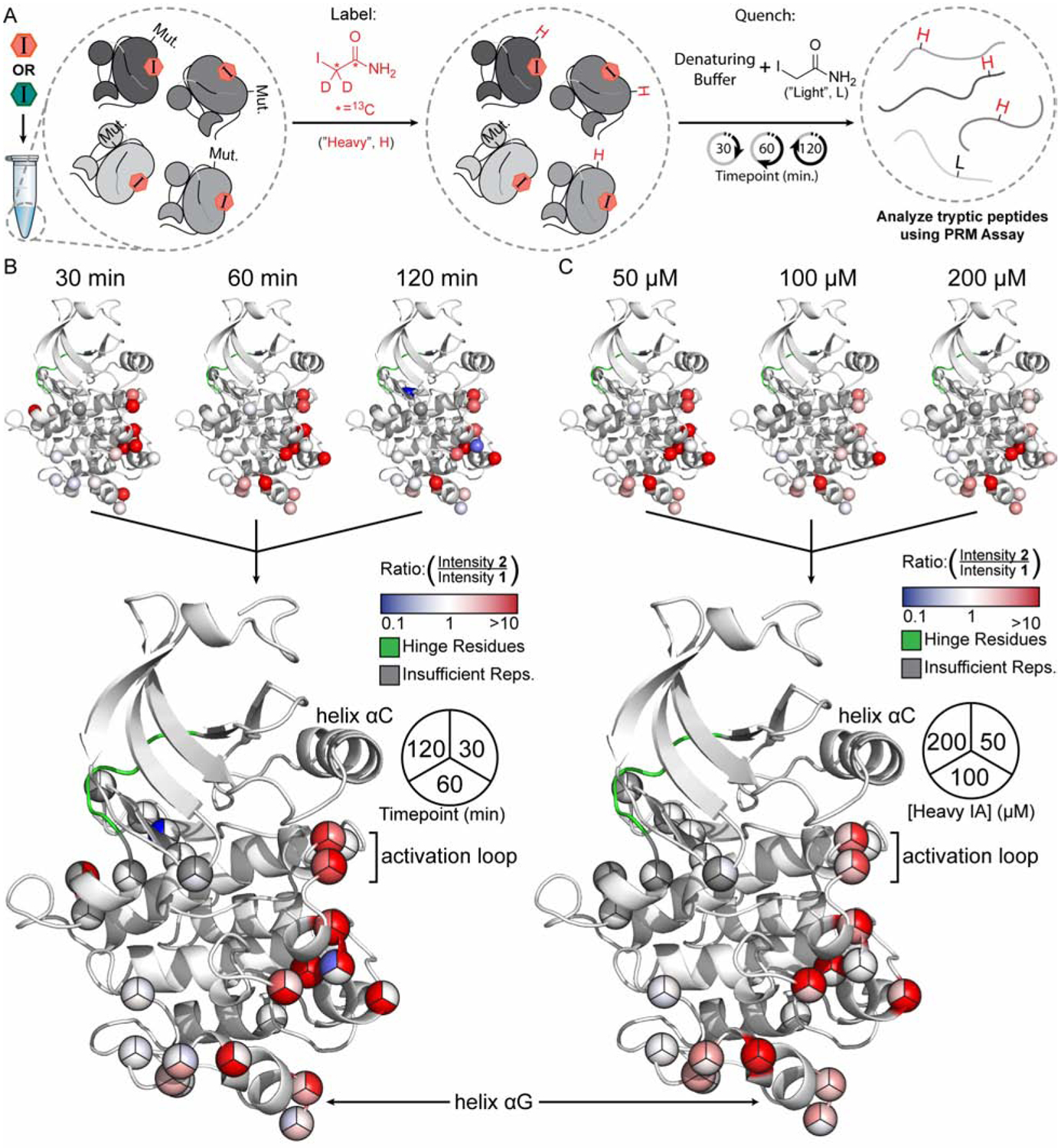
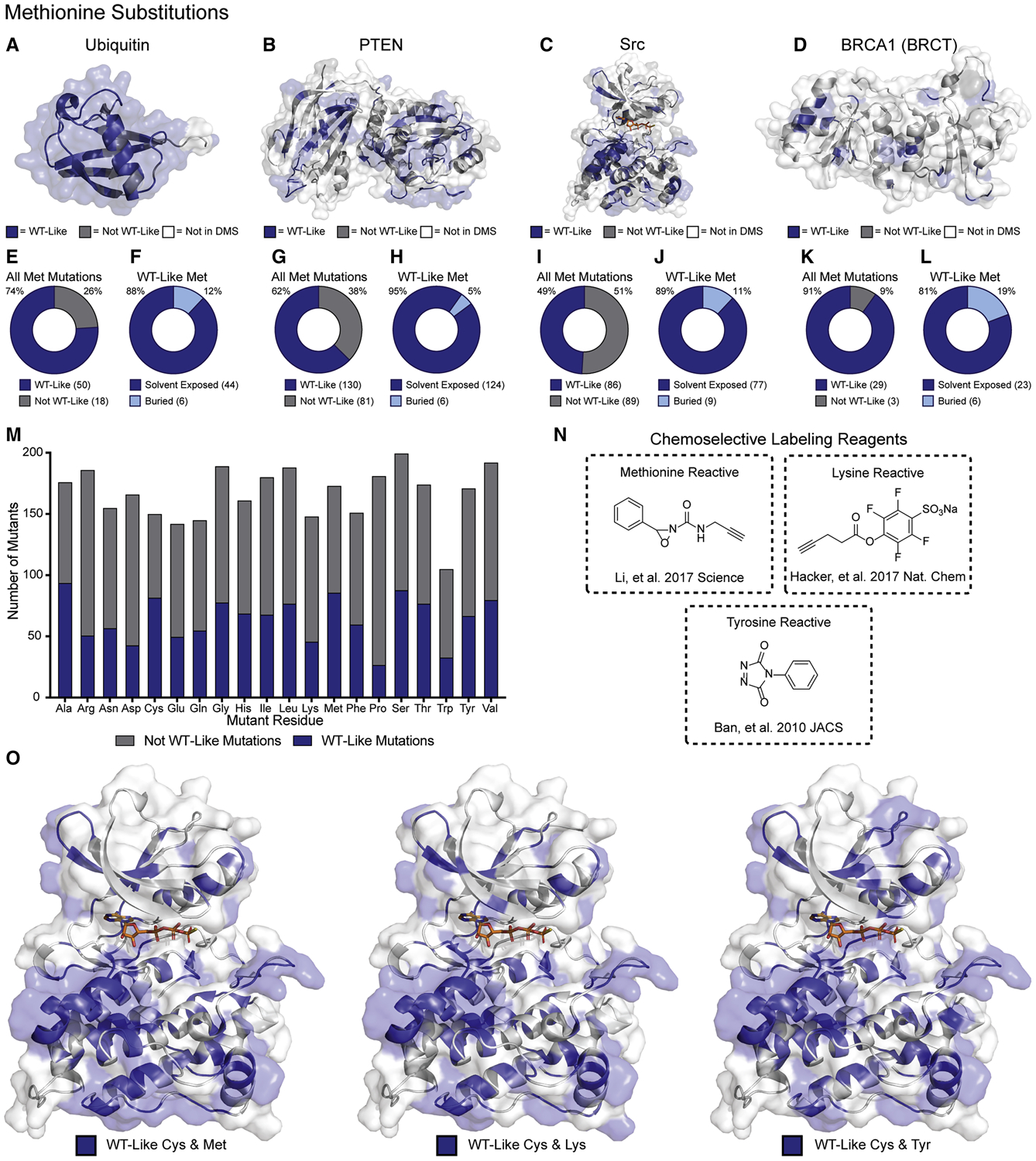
References
-
- Ahler E Register AC, Chakraborty S, Fang L, Dieter EM, Sitko KA, Vidadala RSR, Trevillian BM, Golkowski M, Gelman H, Stephany JJ, Rubin AF, Merritt EA, Fowler DM, Maly DJ (2019). A combined approach reveals a regulatory mechanism coupling Src’s kinase activity, localization and phosphotransferase-independent functions. Mol. Cell74, 2, 393–408 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
