

Bacteroides thetaiotaomicron-Infecting Bacteriophage Isolates Inform Sequence-Based Host Range Predictions
- PMID: 32652063
- PMCID: PMC8045012
- DOI: 10.1016/j.chom.2020.06.011
Bacteroides thetaiotaomicron-Infecting Bacteriophage Isolates Inform Sequence-Based Host Range Predictions
Abstract
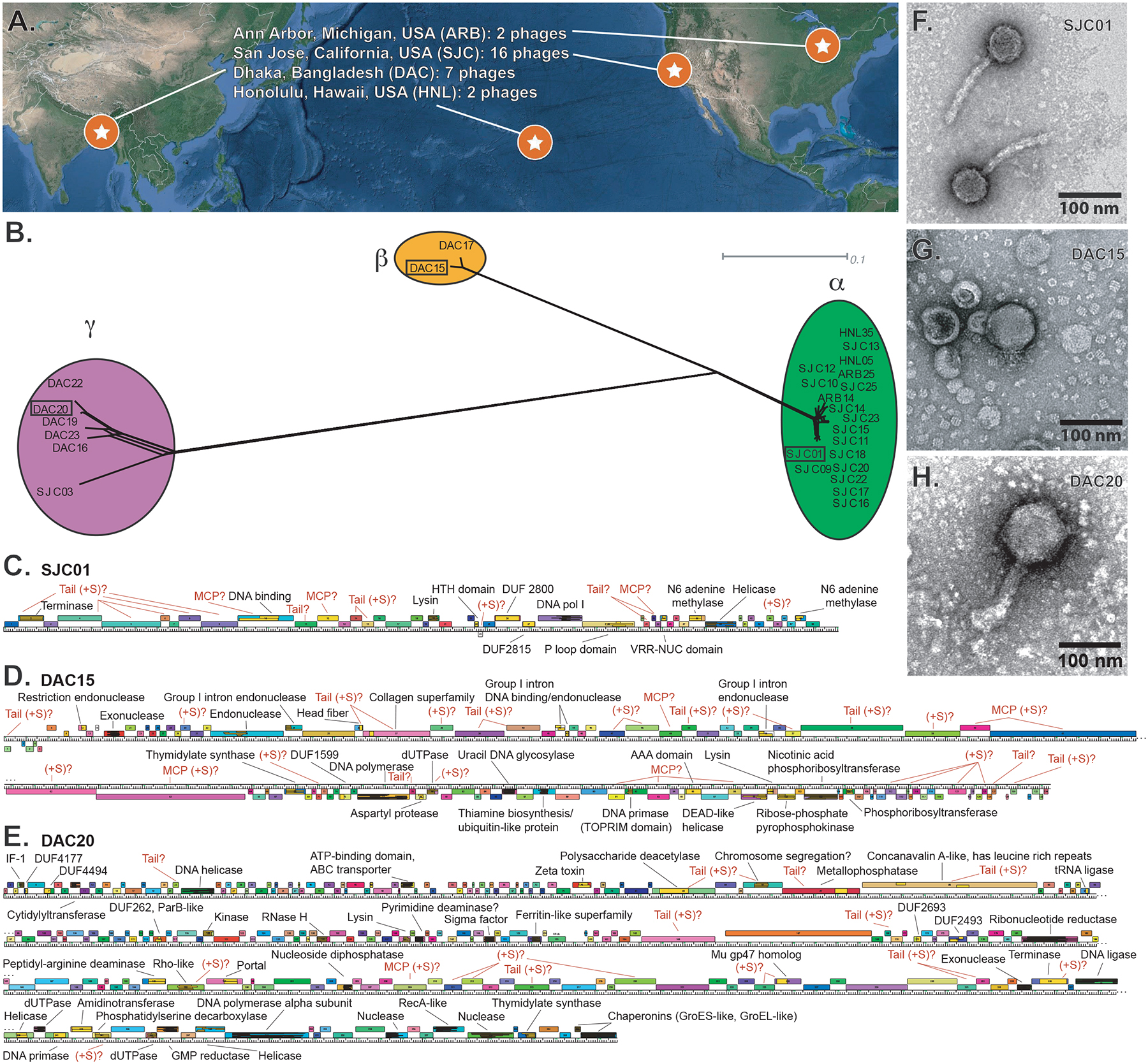
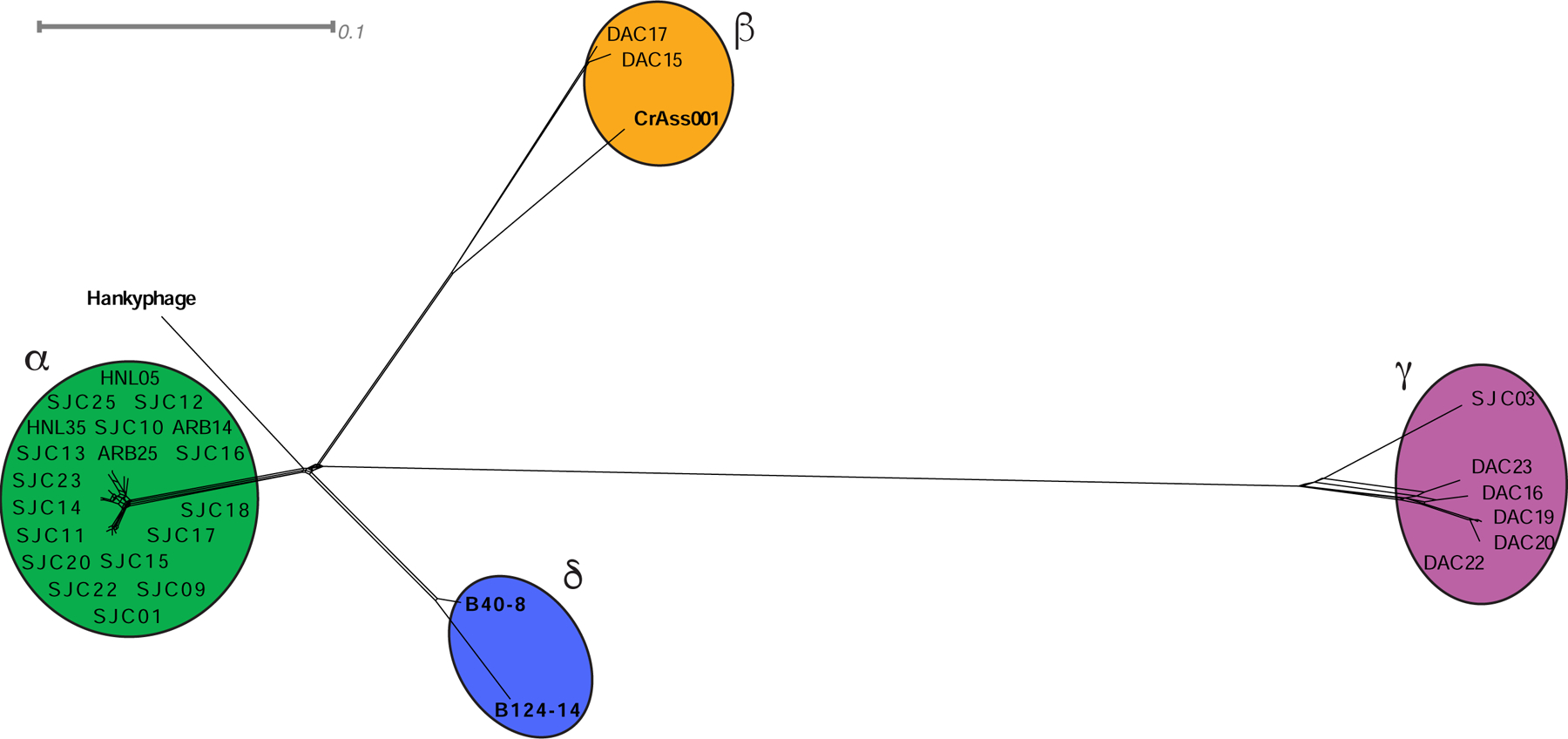
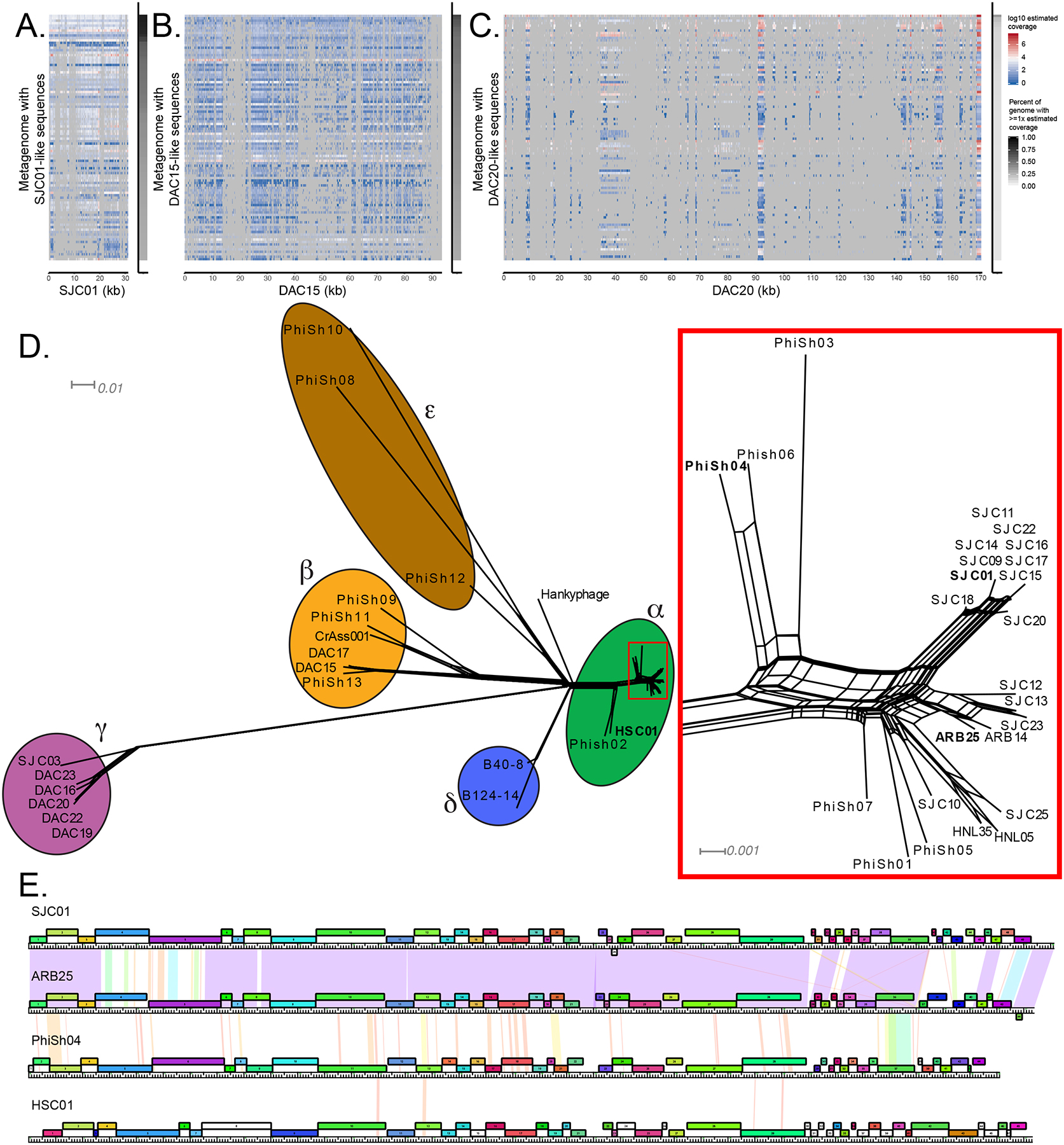
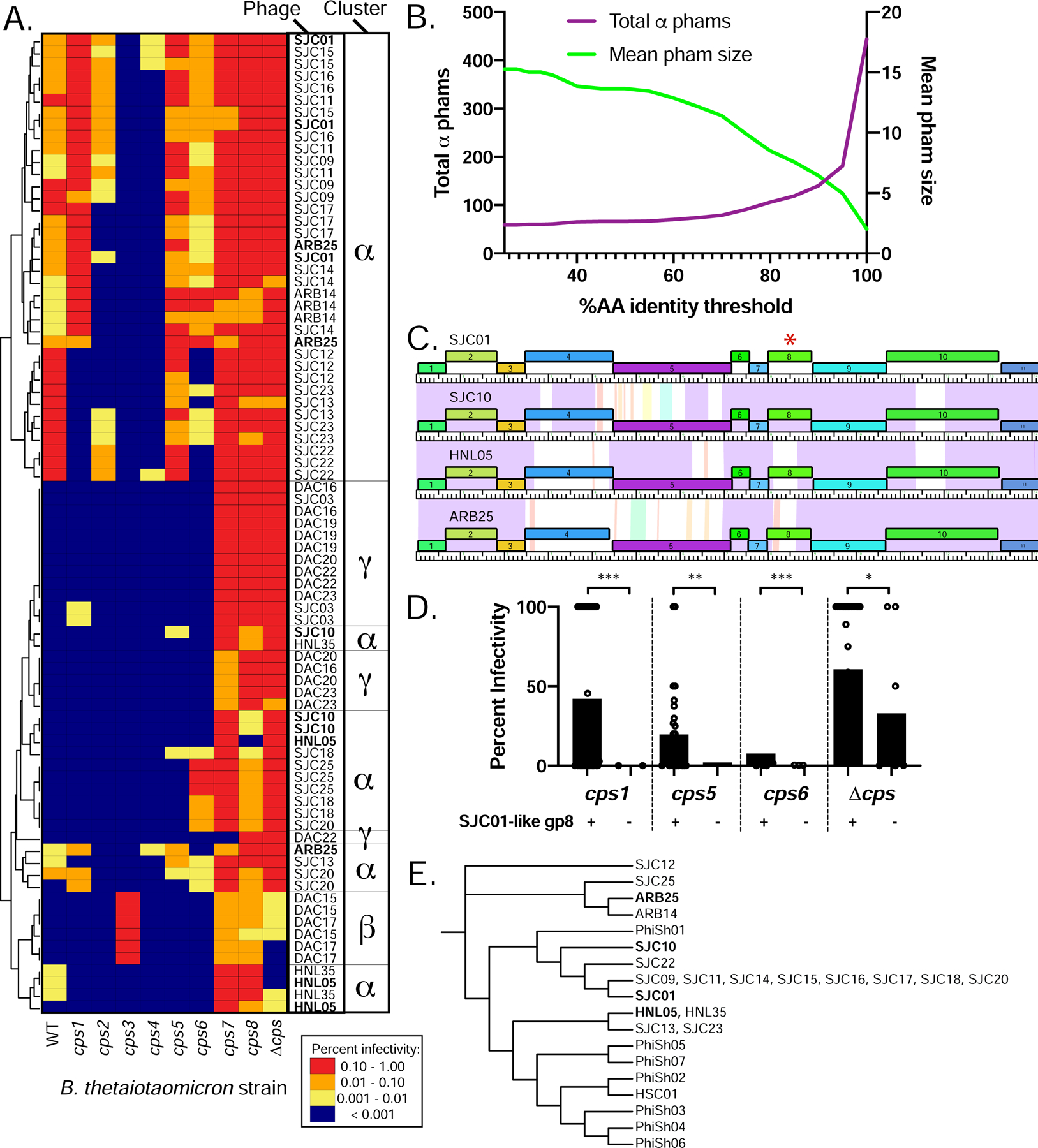
Our emerging view of the gut microbiome largely focuses on bacteria, while less is known about other microbial components, such as bacteriophages (phages). Though phages are abundant in the gut, very few phages have been isolated from this ecosystem. Here, we report the genomes of 27 phages from the United States and Bangladesh that infect the prevalent human gut bacterium Bacteroides thetaiotaomicron. These phages are mostly distinct from previously sequenced phages with the exception of two, which are crAss-like phages. We compare these isolates to existing human gut metagenomes, revealing similarities to previously inferred phages and additional unexplored phage diversity. Finally, we use host tropisms of these phages to identify alleles of phage structural genes associated with infectivity. This work provides a detailed view of the gut's "viral dark matter" and a framework for future efforts to further integrate isolation- and sequencing-focused efforts to understand gut-resident phages.
Keywords: Bacteroides; Bacteroides thetaiotaomicron; bacteriophage; comparative genomics; gut microbiome; metagenomics; microbiota; phage; phage isolation.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
Comment in
-
Phage-Bacteria Associations: Analyze. Match. Develop Therapies.Cell Host Microbe. 2020 Sep 9;28(3):353-355. doi: 10.1016/j.chom.2020.08.009. Cell Host Microbe. 2020. PMID: 32910916
References
-
- BARR JJ, AURO R, FURLAN M, WHITESON KL, ERB ML, POGLIANO J, STOTLAND A, WOLKOWICZ R, CUTTING AS, DORAN KS, SALAMON P, YOULE M & ROHWER F 2013. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci U S A, 110, 10771–6. DOI: 10.1073/pnas.1305923110 - DOI - PMC - PubMed
-
- BIN JANG H, BOLDUC B, ZABLOCKI O, KUHN JH, ROUX S, ADRIAENSSENS EM, BRISTER JR, KROPINSKI AM, KRUPOVIC M, LAVIGNE R, TURNER D & SULLIVAN MB 2019. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat Biotechnol, 37, 632–639. DOI: 10.1038/s41587-019-0100-8 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
