

A Family With a Complex Phenotype Caused by Two Different Rare Metabolic Disorders: GLUT1 and Very-Long-Chain Fatty Acid Dehydrogenase (VLCAD) Deficiencies
- PMID: 32655480
- PMCID: PMC7324651
- DOI: 10.3389/fneur.2020.00514
A Family With a Complex Phenotype Caused by Two Different Rare Metabolic Disorders: GLUT1 and Very-Long-Chain Fatty Acid Dehydrogenase (VLCAD) Deficiencies
Abstract
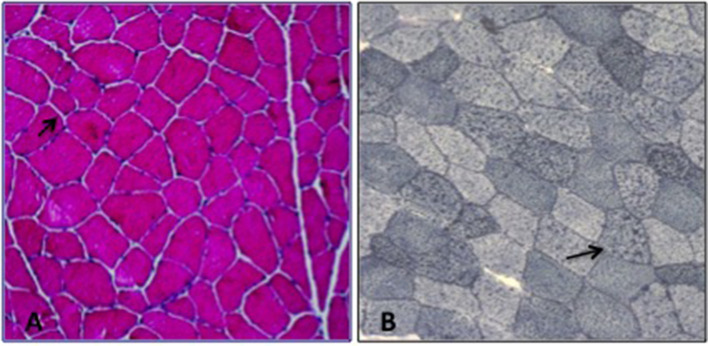
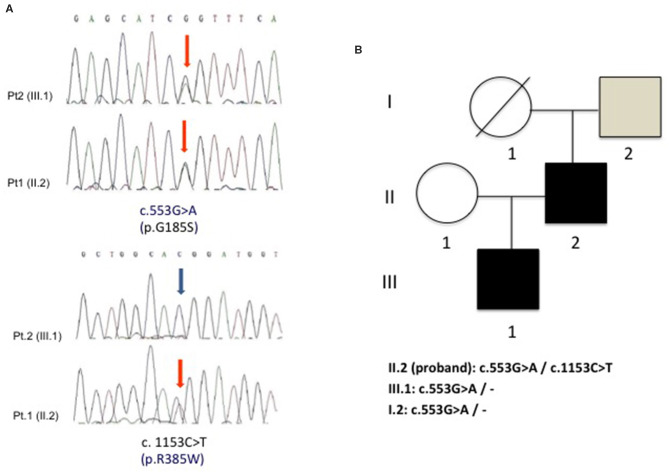
GLUT1 Deficiency Syndrome (GLUT1-DS) is a rare and potentially treatable neurometabolic condition, caused by a reduced glucose transport into the brain and clinically characterized by an epileptic encephalopathy with movement disorders. A wide inter-intrafamilial phenotypic variability has been reported. Very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency is an inherited metabolic disorder of mitochondrial long-chain fatty acid oxidation (FAO) with also a variable age of onset and clinical presentation including cardiomyopathy, hypoketotic hypoglycemia, and liver disease. Sometimes, VLCAD manifests later with a prevalent muscle involvement characterized by exercise intolerance and recurrent rhabdomyolysis. We report a 40-year-old man with mild mental retardation and sporadic choreo-athetoid movements, who complained of recurrent episodes of rhabdomyolysis triggered by exercise or fasting since his twenties. His 15-year-old son had a psychomotor developmental delay with episodes of drowsiness mainly at fasting and exercise-induced choreo-athetoid movements but no history of pigmenturia. Clinical and laboratory findings in the son suggested a diagnosis of GLUT1-DS confirmed by SCL2A1 genetic analysis that revealed a heterozygous mutation c.997C>T (p.R333W) that was also found in the proband. However, the presence in the latter of recurrent exercise-induced rhabdomyolysis, never reported in GLUT1-DS, implied a second metabolic disorder. Increased plasma C14:1-carnitine levels and the identification of two known heterozygous mutations c. 553G>A (p.G185S) and c.1153C>T (p.R385W) in ACADVL confirmed the additional diagnosis of VLCAD deficiency in the proband. Nowadays, there is an increasing evidence of "double trouble" cases of genetic origin. Consequently, when atypical features accompany a known phenotype, associated comorbidities should be considered.
Keywords: GLUT1-DS; VLCAD; genetic “double-trouble”; metabolic myopathy; rhabdomyolysis.
Copyright © 2020 Musumeci, Ferlazzo, Rodolico, Gambardella, Gagliardi, Aguglia and Toscano.
Figures
Similar articles
-
A heterozygous missense mutation in adolescent-onset very long-chain acyl-CoA dehydrogenase deficiency with exercise-induced rhabdomyolysis.Tohoku J Exp Med. 2015 Apr;235(4):305-10. doi: 10.1620/tjem.235.305. Tohoku J Exp Med. 2015. PMID: 25843429
-
Diagnosis of very long chain acyl-dehydrogenase deficiency from an infant's newborn screening card.Pediatrics. 2001 Jul;108(1):E19. doi: 10.1542/peds.108.1.e19. Pediatrics. 2001. PMID: 11433098
-
Diagnostic assessment and long-term follow-up of 13 patients with Very Long-Chain Acyl-Coenzyme A dehydrogenase (VLCAD) deficiency.Neuromuscul Disord. 2009 May;19(5):324-9. doi: 10.1016/j.nmd.2009.02.007. Epub 2009 Mar 26. Neuromuscul Disord. 2009. PMID: 19327992
-
[Very-long-chain acyl-CoA dehydrogenase deficiency presenting with recurrent rhabdomyolysis in an adult].Rinsho Shinkeigaku. 2003 May;43(5):253-7. Rinsho Shinkeigaku. 2003. PMID: 12931630 Review. Japanese.
-
New genetic defects in mitochondrial fatty acid oxidation and carnitine deficiency.Adv Pediatr. 1987;34:59-88. Adv Pediatr. 1987. PMID: 3318304 Review.
Cited by
-
Multi-Omics Integration Analysis Pinpoint Proteins Influencing Brain Structure and Function: Toward Drug Targets and Neuroimaging Biomarkers for Neuropsychiatric Disorders.Int J Mol Sci. 2024 Aug 25;25(17):9223. doi: 10.3390/ijms25179223. Int J Mol Sci. 2024. PMID: 39273172 Free PMC article.
-
Phenotypic and Genotypic Spectrum of Early-Onset Developmental and Epileptic Encephalopathies-Data from a Romanian Cohort.Genes (Basel). 2022 Jul 15;13(7):1253. doi: 10.3390/genes13071253. Genes (Basel). 2022. PMID: 35886038 Free PMC article.
-
Diagnosis and treatment recommendations for glucose transporter 1 deficiency syndrome.World J Pediatr. 2025 Feb;21(2):149-158. doi: 10.1007/s12519-024-00864-5. Epub 2025 Jan 2. World J Pediatr. 2025. PMID: 39745620 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Miscellaneous