

The DNA methylation landscape of advanced prostate cancer
- PMID: 32661416
- PMCID: PMC7454228
- DOI: 10.1038/s41588-020-0648-8
The DNA methylation landscape of advanced prostate cancer
Abstract
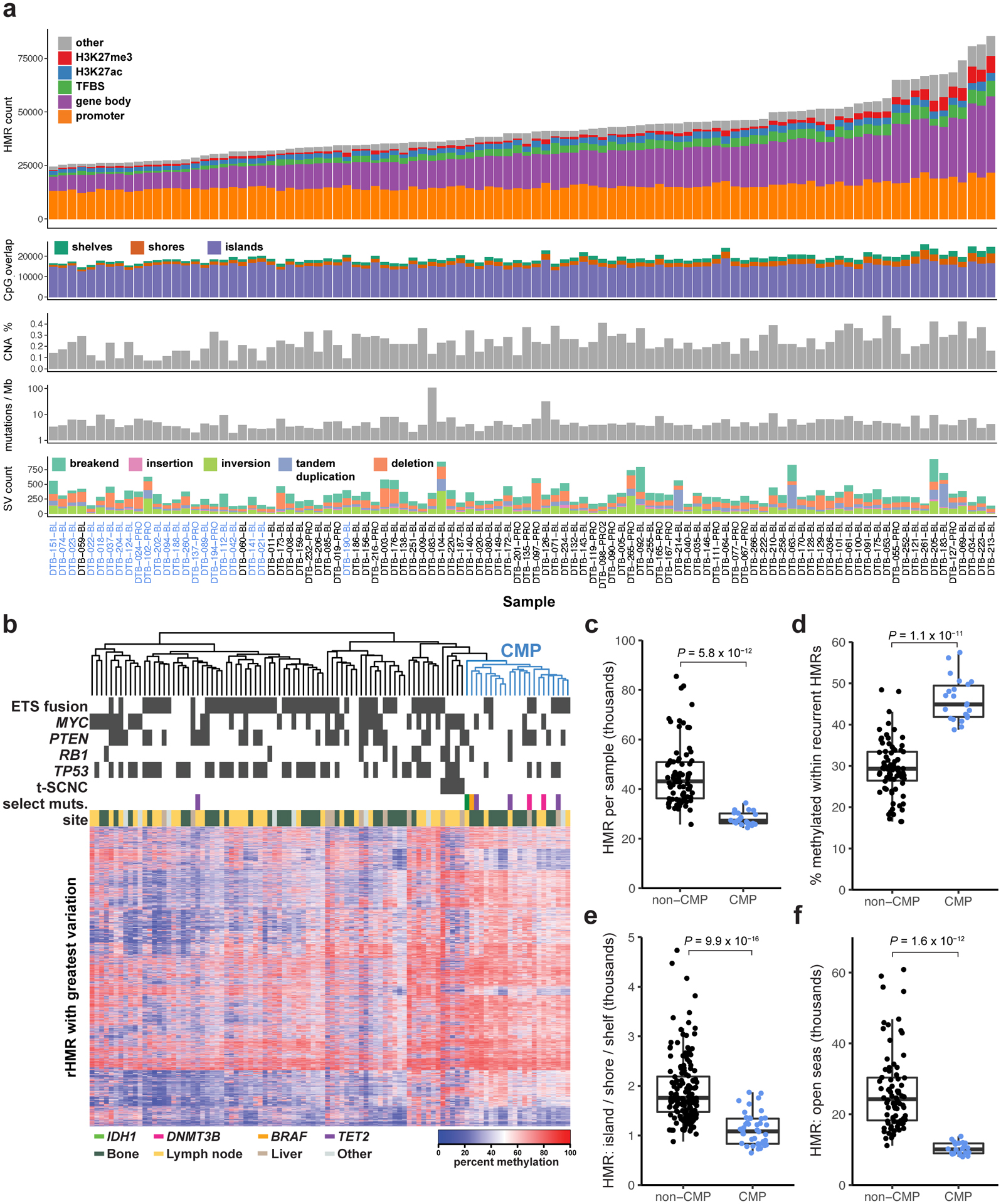
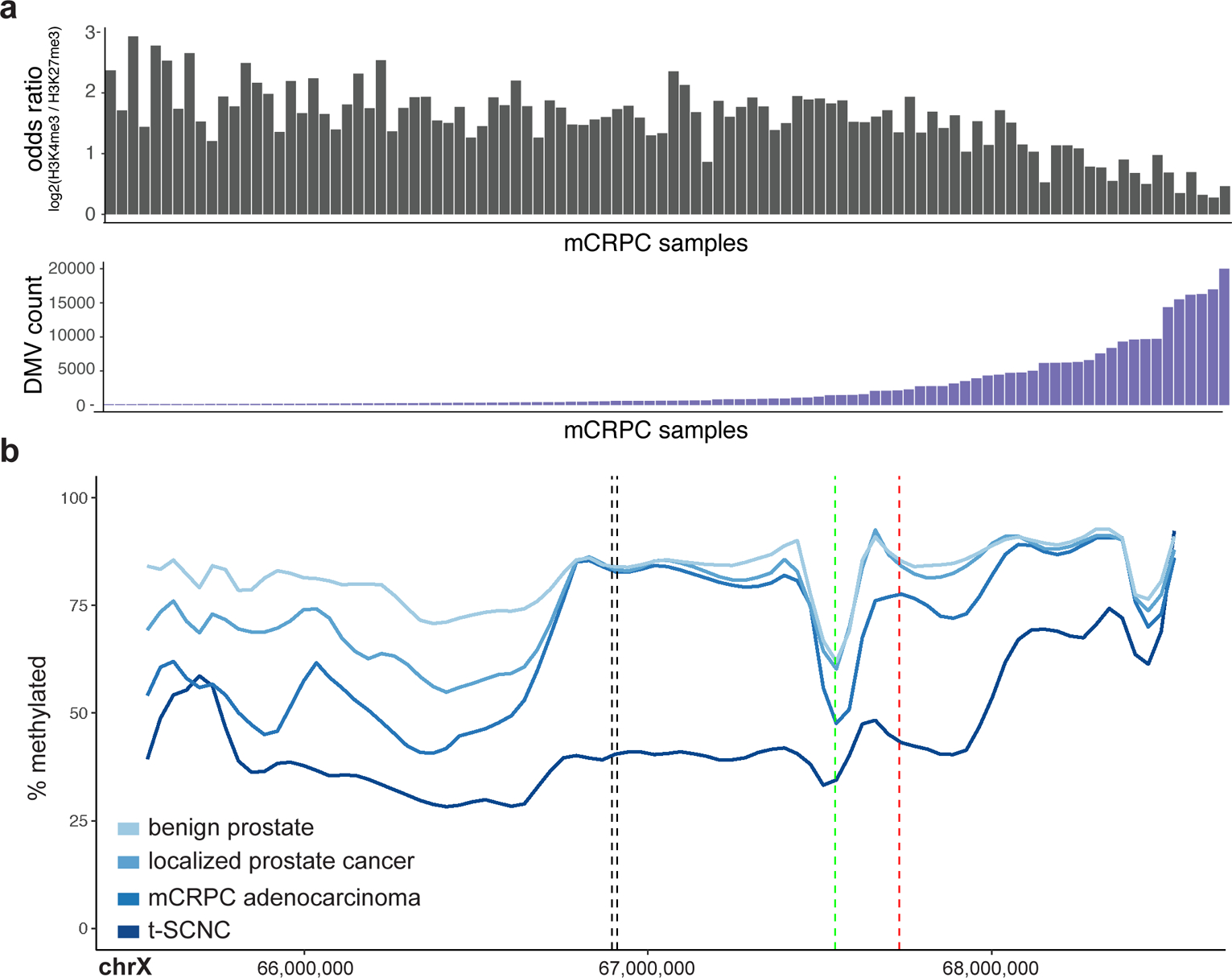
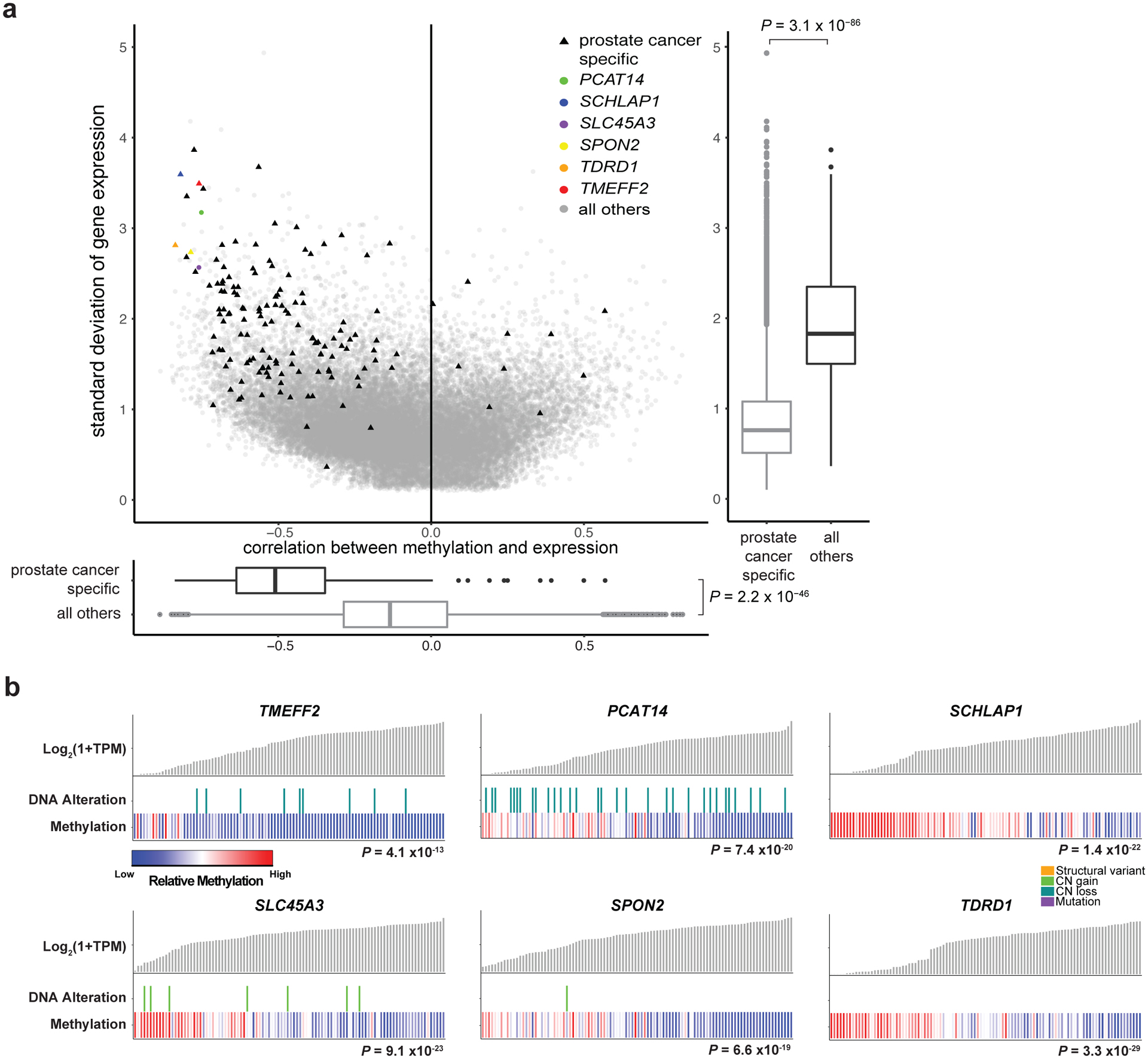
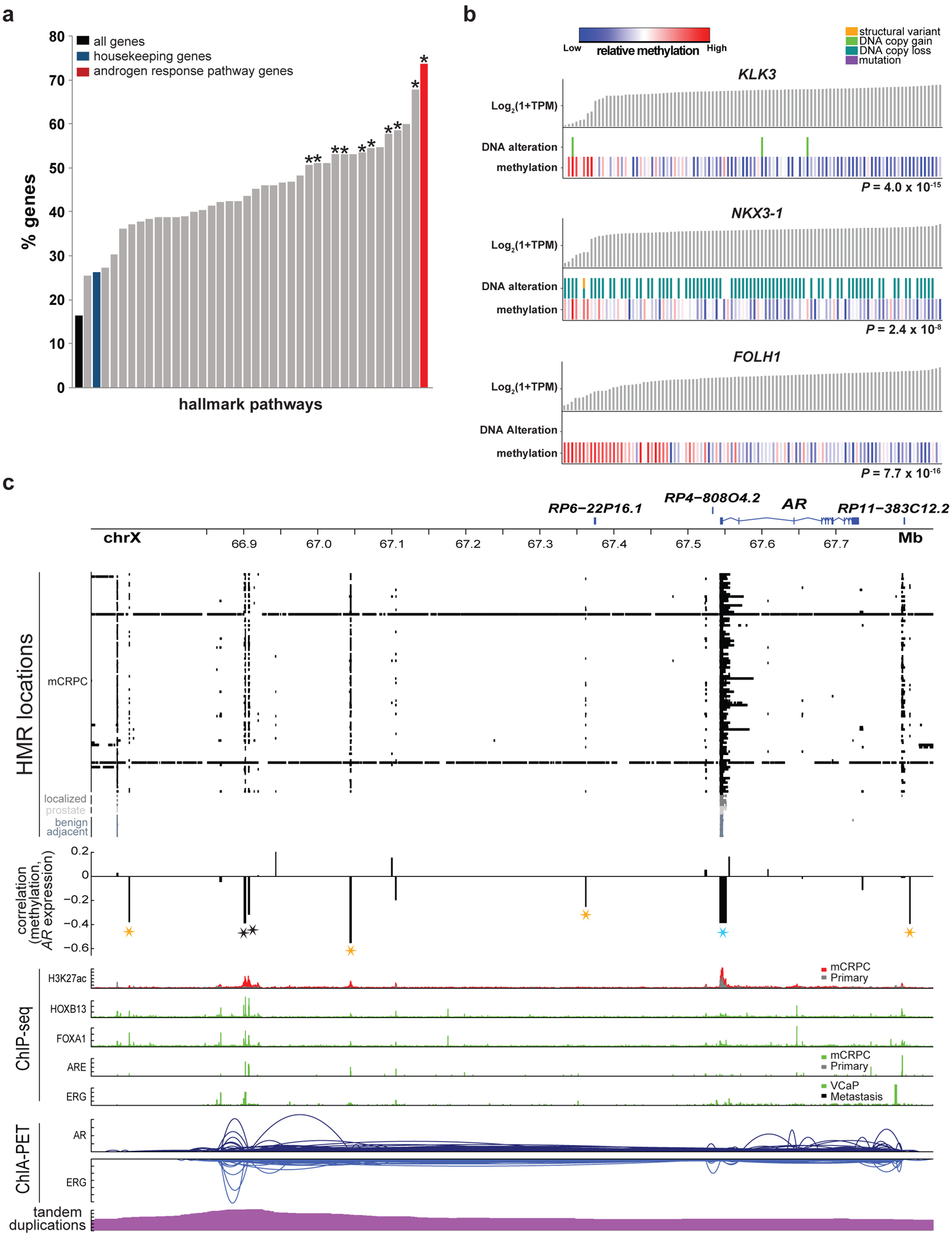
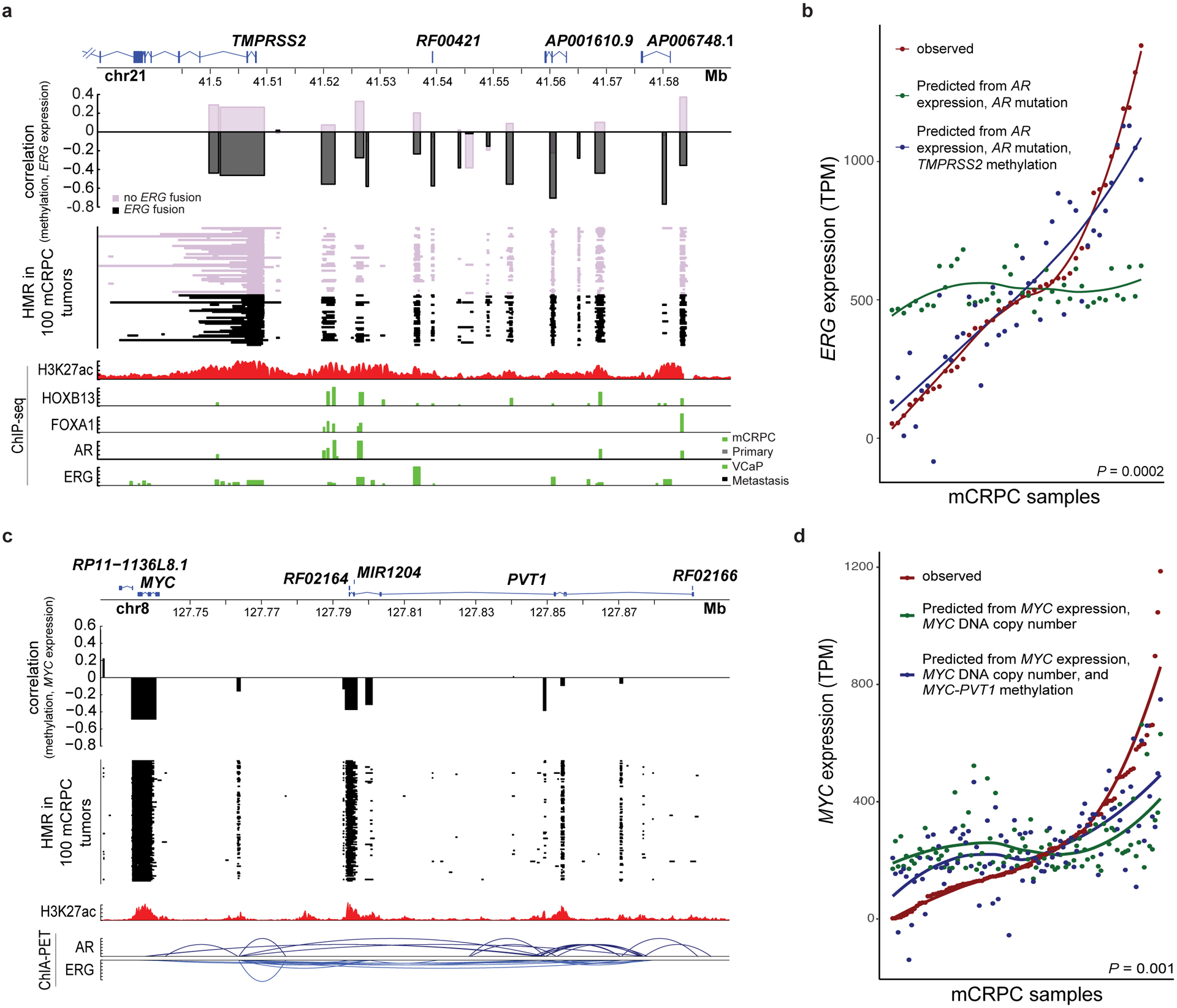
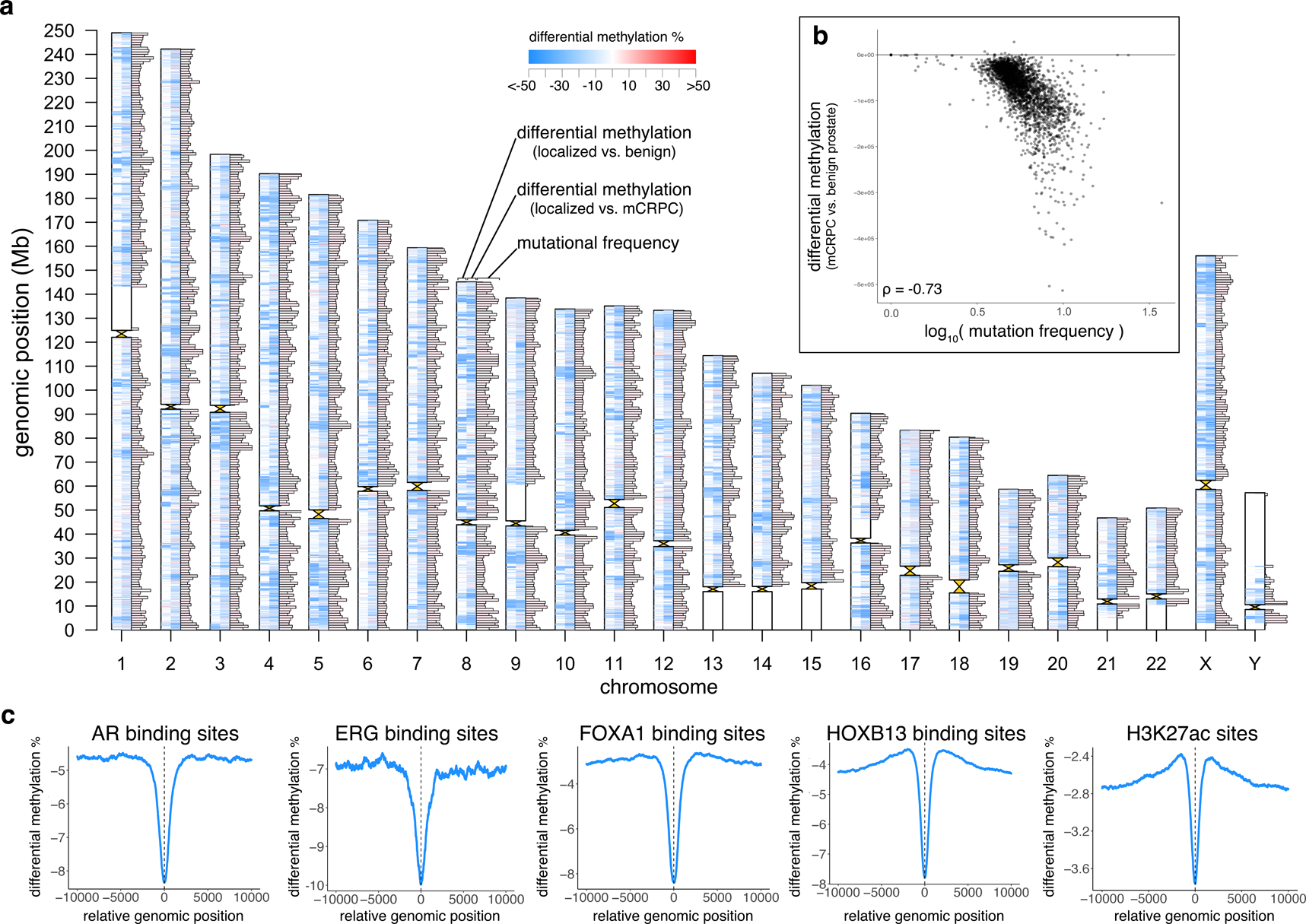
Although DNA methylation is a key regulator of gene expression, the comprehensive methylation landscape of metastatic cancer has never been defined. Through whole-genome bisulfite sequencing paired with deep whole-genome and transcriptome sequencing of 100 castration-resistant prostate metastases, we discovered alterations affecting driver genes that were detectable only with integrated whole-genome approaches. Notably, we observed that 22% of tumors exhibited a novel epigenomic subtype associated with hypermethylation and somatic mutations in TET2, DNMT3B, IDH1 and BRAF. We also identified intergenic regions where methylation is associated with RNA expression of the oncogenic driver genes AR, MYC and ERG. Finally, we showed that differential methylation during progression preferentially occurs at somatic mutational hotspots and putative regulatory regions. This study is a large integrated study of whole-genome, whole-methylome and whole-transcriptome sequencing in metastatic cancer that provides a comprehensive overview of the important regulatory role of methylation in metastatic castration-resistant prostate cancer.
Figures
References
-
- Jones PA & Baylin SB The fundamental role of epigenetic events in cancer. Nat Rev Genet 3, 415–28 (2002). - PubMed
-
- Okano M, Bell DW, Haber DA & Li E DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–57 (1999). - PubMed
-
- Skvortsova K et al. DNA Hypermethylation Encroachment at CpG Island Borders in Cancer Is Predisposed by H3K4 Monomethylation Patterns. Cancer Cell 35, 297–314 e8 (2019). - PubMed
Publication types
MeSH terms
Grants and funding
- U10 CA180868/CA/NCI NIH HHS/United States
- P30 CA093373/CA/NCI NIH HHS/United States
- R01 CA227025/CA/NCI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 CA230516/CA/NCI NIH HHS/United States
- DP2 CA239597/CA/NCI NIH HHS/United States
- R00 CA204602/CA/NCI NIH HHS/United States
- P50 CA211015/CA/NCI NIH HHS/United States
- P50 CA186786/CA/NCI NIH HHS/United States
- K99 CA204602/CA/NCI NIH HHS/United States
- U01 CA214194/CA/NCI NIH HHS/United States
- R01 CA174777/CA/NCI NIH HHS/United States
- R35 CA231996/CA/NCI NIH HHS/United States
- U01 CA214170/CA/NCI NIH HHS/United States
- R01 CA208258/CA/NCI NIH HHS/United States
- P50 CA092131/CA/NCI NIH HHS/United States
- P30 CA016042/CA/NCI NIH HHS/United States
- R01 CA240582/CA/NCI NIH HHS/United States
- P30 ES017885/ES/NIEHS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
