

Machine learning classification can reduce false positives in structure-based virtual screening
- PMID: 32669436
- PMCID: PMC7414157
- DOI: 10.1073/pnas.2000585117
Machine learning classification can reduce false positives in structure-based virtual screening
Abstract
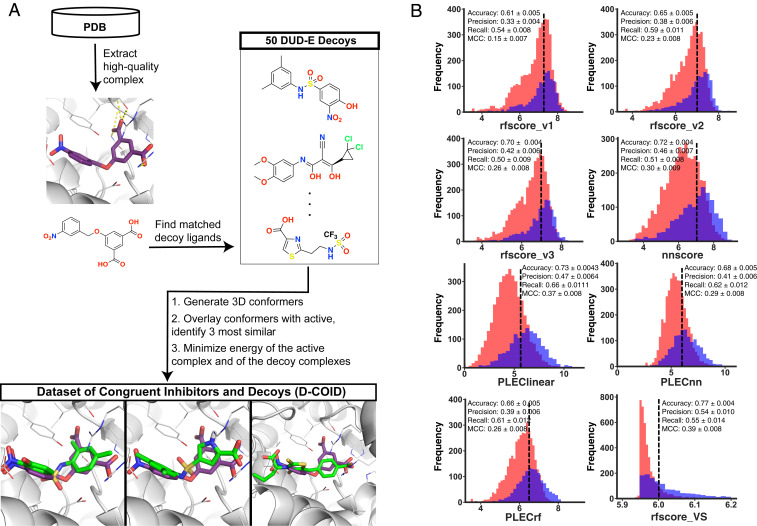
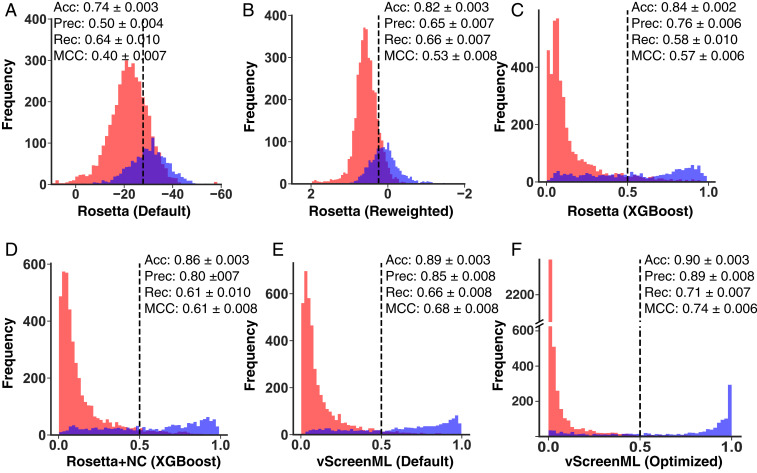
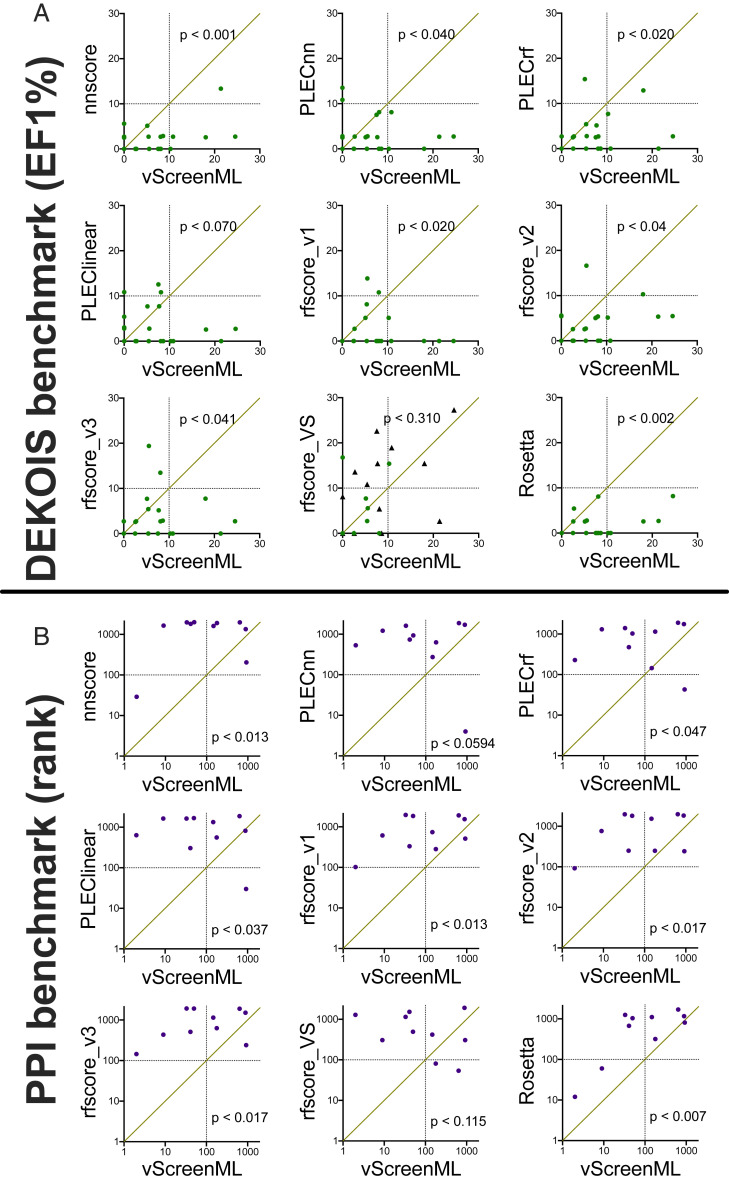
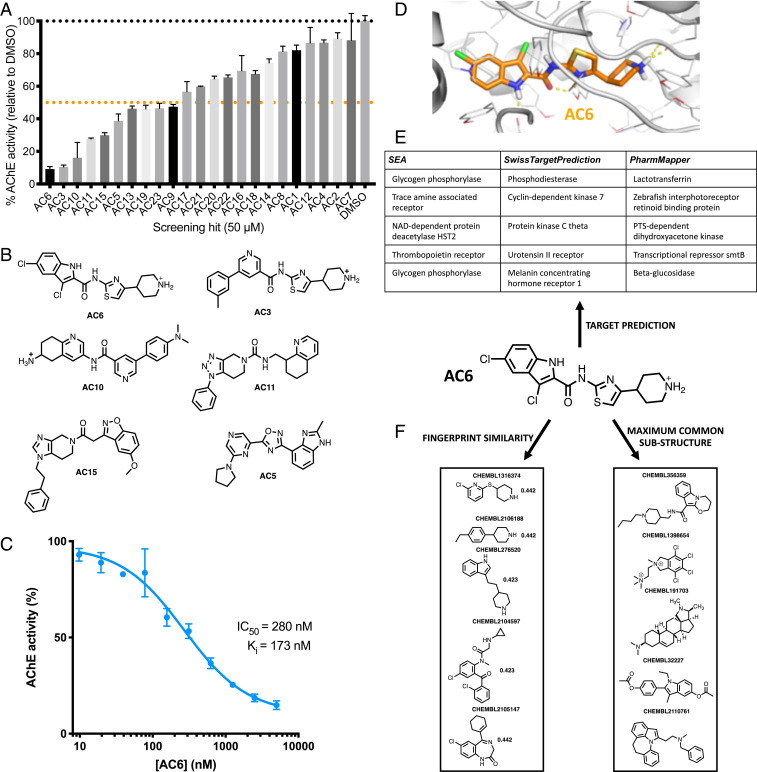
With the recent explosion in the size of libraries available for screening, virtual screening is positioned to assume a more prominent role in early drug discovery's search for active chemical matter. In typical virtual screens, however, only about 12% of the top-scoring compounds actually show activity when tested in biochemical assays. We argue that most scoring functions used for this task have been developed with insufficient thoughtfulness into the datasets on which they are trained and tested, leading to overly simplistic models and/or overtraining. These problems are compounded in the literature because studies reporting new scoring methods have not validated their models prospectively within the same study. Here, we report a strategy for building a training dataset (D-COID) that aims to generate highly compelling decoy complexes that are individually matched to available active complexes. Using this dataset, we train a general-purpose classifier for virtual screening (vScreenML) that is built on the XGBoost framework. In retrospective benchmarks, our classifier shows outstanding performance relative to other scoring functions. In a prospective context, nearly all candidate inhibitors from a screen against acetylcholinesterase show detectable activity; beyond this, 10 of 23 compounds have IC50 better than 50 μM. Without any medicinal chemistry optimization, the most potent hit has IC50 280 nM, corresponding to Ki of 173 nM. These results support using the D-COID strategy for training classifiers in other computational biology tasks, and for vScreenML in virtual screening campaigns against other protein targets. Both D-COID and vScreenML are freely distributed to facilitate such efforts.
Keywords: machine learning classifier; protein–ligand complex; structure-based drug design; virtual screening.
Copyright © 2020 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
Similar articles
-
vScreenML v2.0: Improved Machine Learning Classification for Reducing False Positives in Structure-Based Virtual Screening.Int J Mol Sci. 2024 Nov 18;25(22):12350. doi: 10.3390/ijms252212350. Int J Mol Sci. 2024. PMID: 39596415 Free PMC article.
-
Structure-based virtual screening of vast chemical space as a starting point for drug discovery.Curr Opin Struct Biol. 2024 Aug;87:102829. doi: 10.1016/j.sbi.2024.102829. Epub 2024 Jun 6. Curr Opin Struct Biol. 2024. PMID: 38848655 Review.
-
Improving virtual screening predictive accuracy of Human kallikrein 5 inhibitors using machine learning models.Comput Biol Chem. 2017 Aug;69:110-119. doi: 10.1016/j.compbiolchem.2017.05.007. Epub 2017 May 29. Comput Biol Chem. 2017. PMID: 28601761
-
Automated Inference of Chemical Discriminants of Biological Activity.Methods Mol Biol. 2018;1762:307-338. doi: 10.1007/978-1-4939-7756-7_16. Methods Mol Biol. 2018. PMID: 29594779
-
Comparative analysis of machine learning methods in ligand-based virtual screening of large compound libraries.Comb Chem High Throughput Screen. 2009 May;12(4):344-57. doi: 10.2174/138620709788167944. Comb Chem High Throughput Screen. 2009. PMID: 19442064 Review.
Cited by
-
A vending machine for drug-like molecules - automated synthesis of virtual screening hits.Chem Sci. 2022 Oct 28;13(48):14292-14299. doi: 10.1039/d2sc05182f. eCollection 2022 Dec 14. Chem Sci. 2022. PMID: 36545137 Free PMC article.
-
G Protein-Coupled Receptor-Ligand Pose and Functional Class Prediction.Int J Mol Sci. 2024 Jun 22;25(13):6876. doi: 10.3390/ijms25136876. Int J Mol Sci. 2024. PMID: 38999982 Free PMC article.
-
Machine learning-aided scoring of synthesis difficulties for designer chromosomes.Sci China Life Sci. 2023 Jul;66(7):1615-1625. doi: 10.1007/s11427-023-2306-x. Epub 2023 Mar 3. Sci China Life Sci. 2023. PMID: 36881317
-
Essential Dynamics Ensemble Docking for Structure-Based GPCR Drug Discovery.Front Mol Biosci. 2022 Jun 29;9:879212. doi: 10.3389/fmolb.2022.879212. eCollection 2022. Front Mol Biosci. 2022. PMID: 35847975 Free PMC article.
-
Energy-entropy method using multiscale cell correlation to calculate binding free energies in the SAMPL8 host-guest challenge.J Comput Aided Mol Des. 2021 Aug;35(8):911-921. doi: 10.1007/s10822-021-00406-5. Epub 2021 Jul 15. J Comput Aided Mol Des. 2021. PMID: 34264476 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
