

Skp2 dictates cell cycle-dependent metabolic oscillation between glycolysis and TCA cycle
- PMID: 32669607
- PMCID: PMC7852548
- DOI: 10.1038/s41422-020-0372-z
Skp2 dictates cell cycle-dependent metabolic oscillation between glycolysis and TCA cycle
Erratum in
-
Author Correction: Skp2 dictates cell cycle-dependent metabolic oscillation between glycolysis and TCA cycle.Cell Res. 2021 Jan;31(1):104. doi: 10.1038/s41422-020-00406-4. Cell Res. 2021. PMID: 32859994 Free PMC article. No abstract available.
Abstract
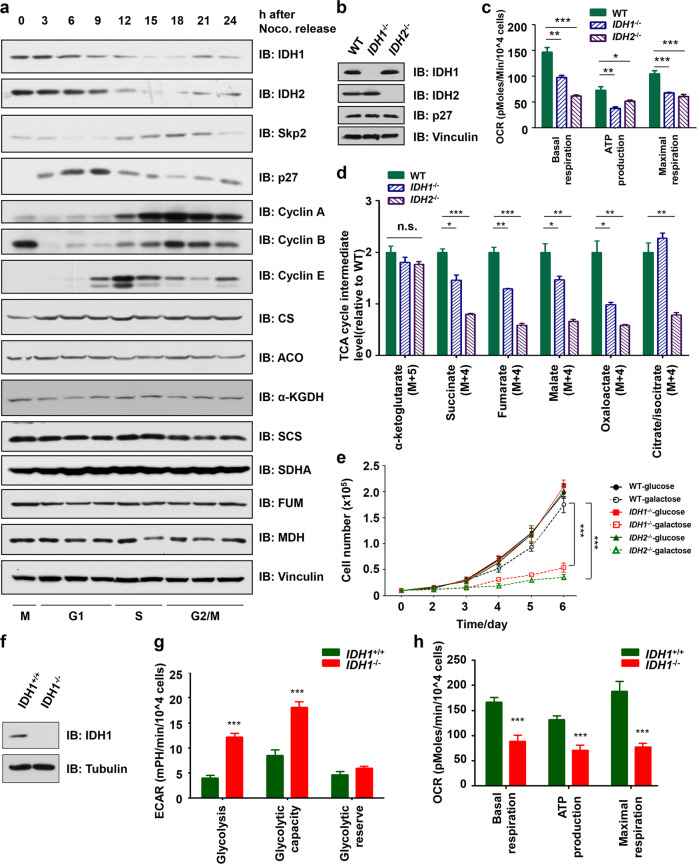
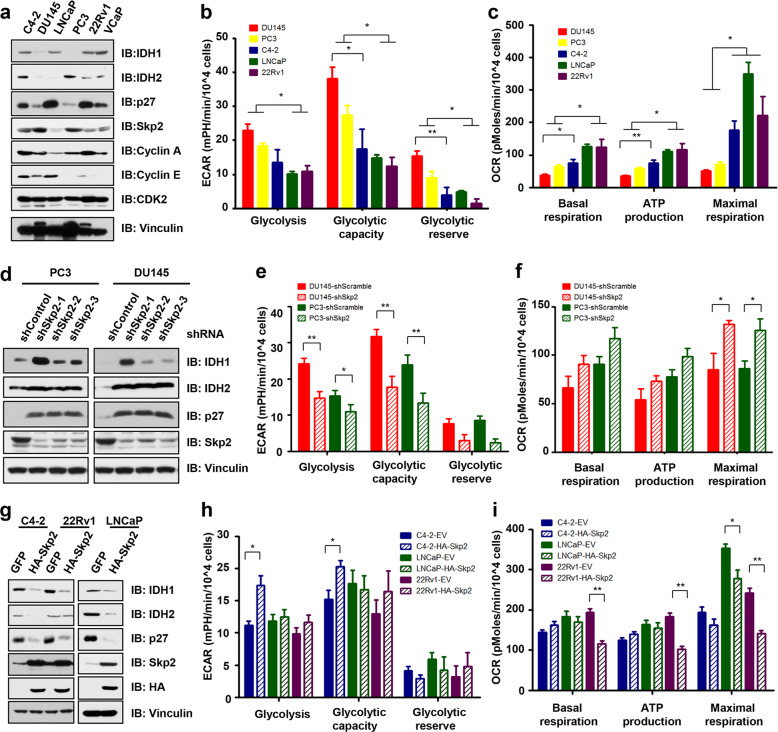
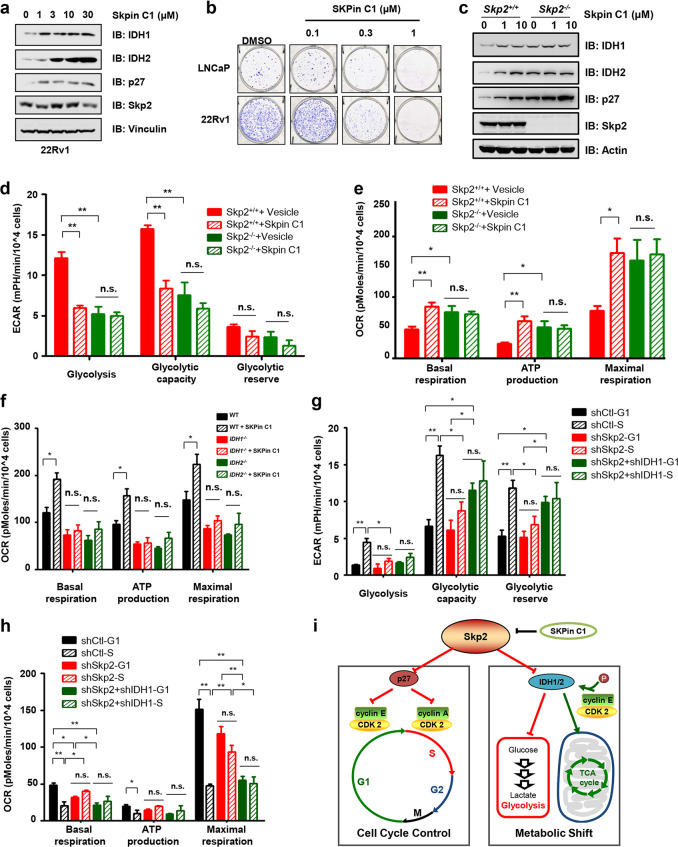
Whether glucose is predominantly metabolized via oxidative phosphorylation or glycolysis differs between quiescent versus proliferating cells, including tumor cells. However, how glucose metabolism is coordinated with cell cycle in mammalian cells remains elusive. Here, we report that mammalian cells predominantly utilize the tricarboxylic acid (TCA) cycle in G1 phase, but prefer glycolysis in S phase. Mechanistically, coupling cell cycle with metabolism is largely achieved by timely destruction of IDH1/2, key TCA cycle enzymes, in a Skp2-dependent manner. As such, depleting SKP2 abolishes cell cycle-dependent fluctuation of IDH1 protein abundance, leading to reduced glycolysis in S phase. Furthermore, elevated Skp2 abundance in prostate cancer cells destabilizes IDH1 to favor glycolysis and subsequent tumorigenesis. Therefore, our study reveals a mechanistic link between two cancer hallmarks, aberrant cell cycle and addiction to glycolysis, and provides the underlying mechanism for the coupling of metabolic fluctuation with periodic cell cycle in mammalian cells.
Conflict of interest statement
W.W. is a co-founder and consultant of the ReKindle Therapeutics. All other authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
