

Ageing hallmarks exhibit organ-specific temporal signatures
- PMID: 32669715
- PMCID: PMC7757734
- DOI: 10.1038/s41586-020-2499-y
Ageing hallmarks exhibit organ-specific temporal signatures
Abstract
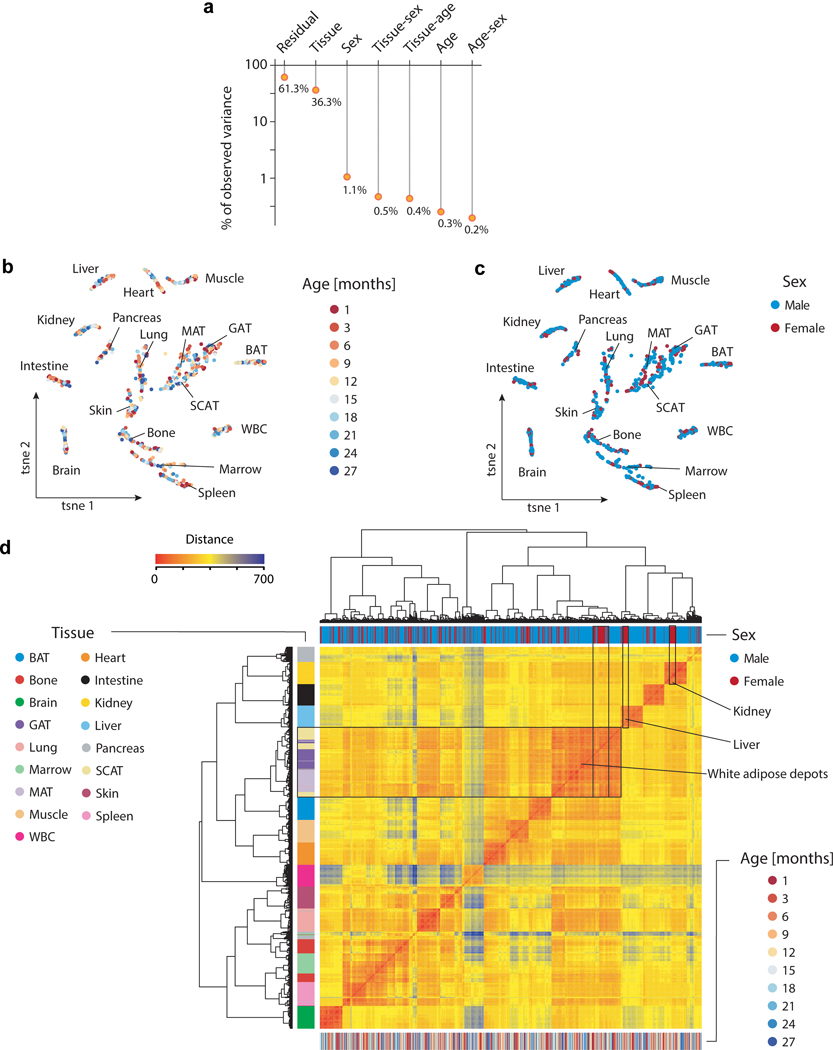
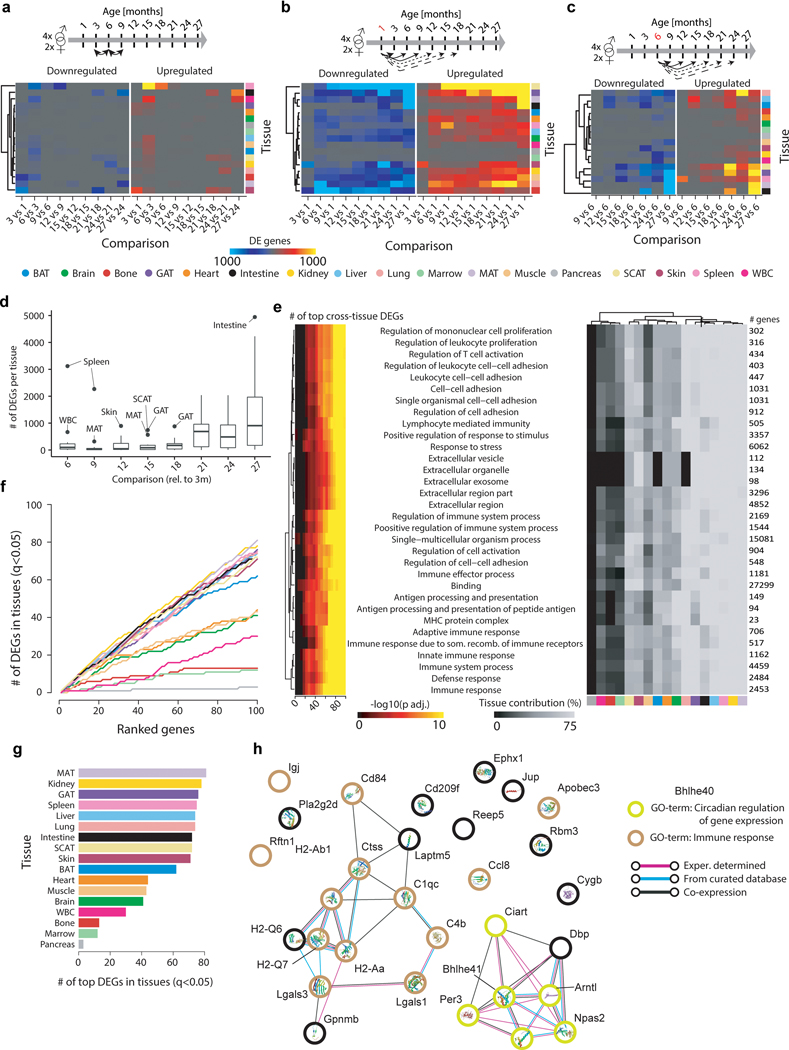
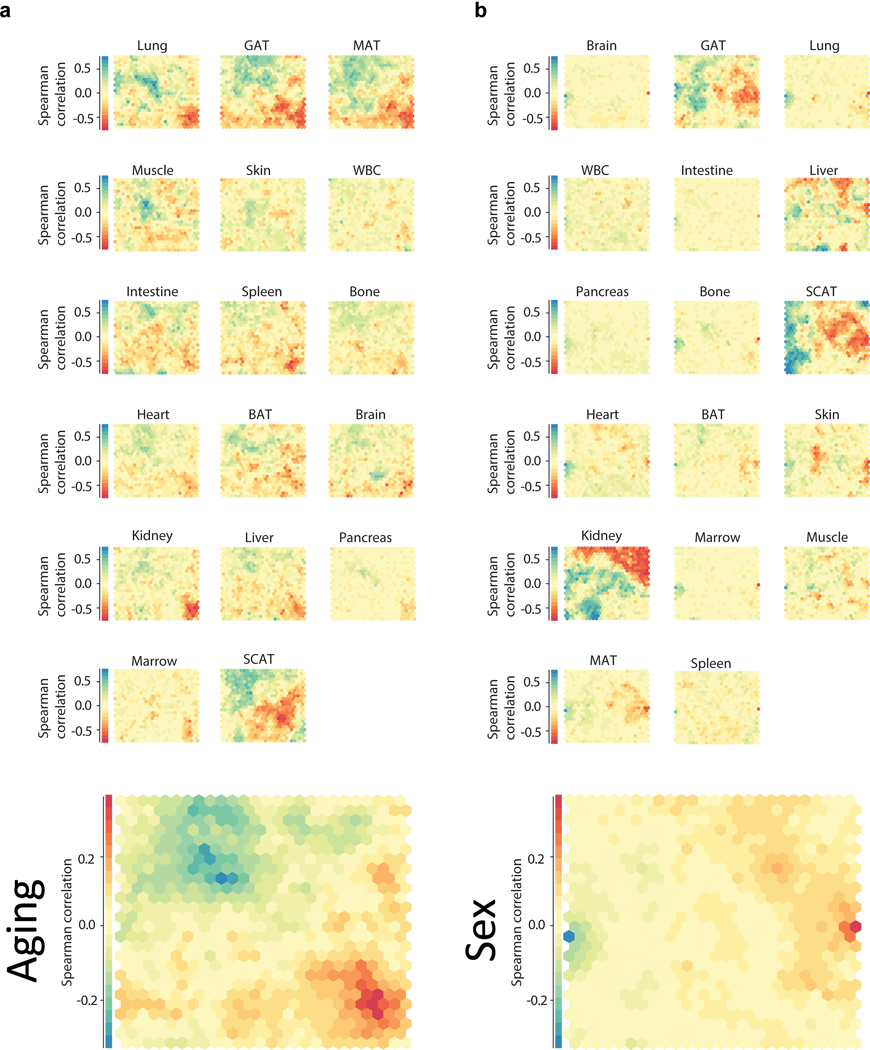
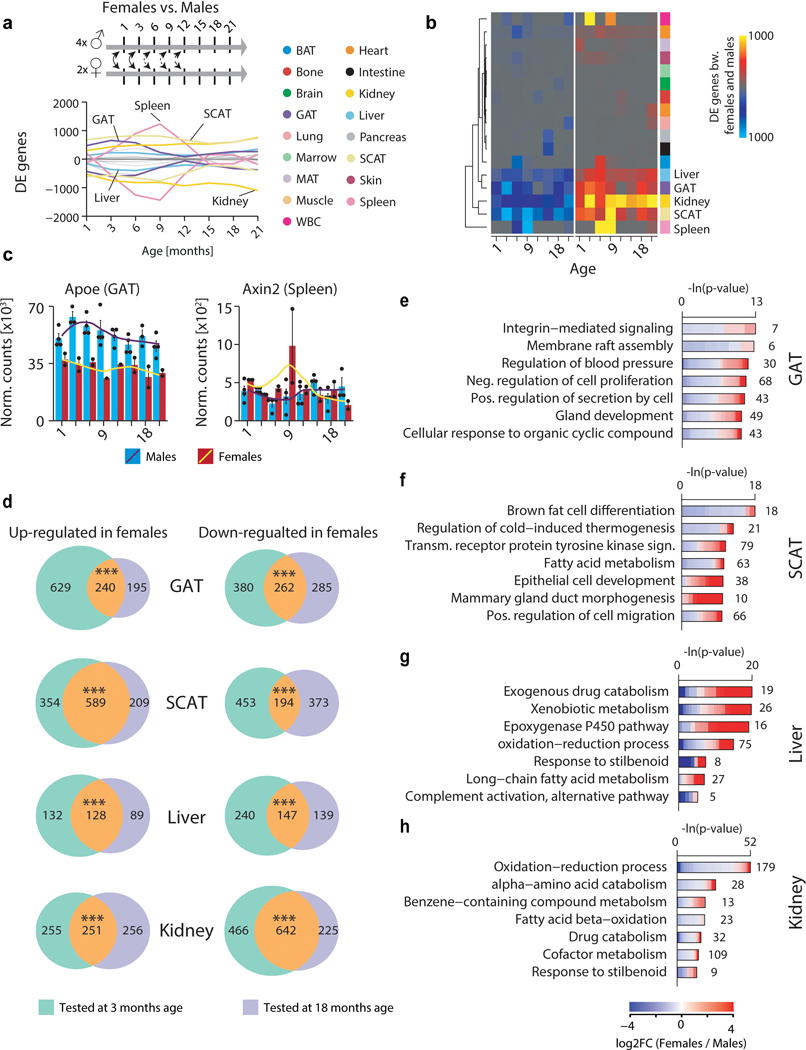
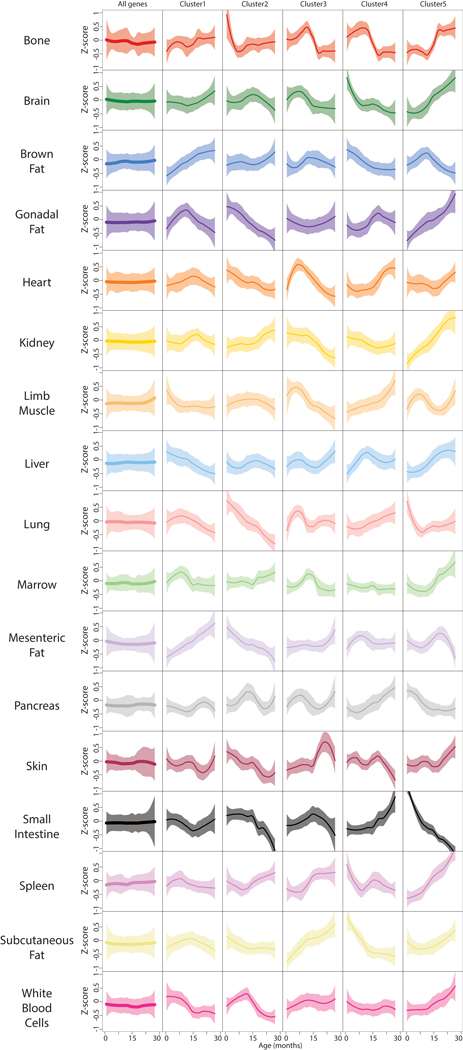
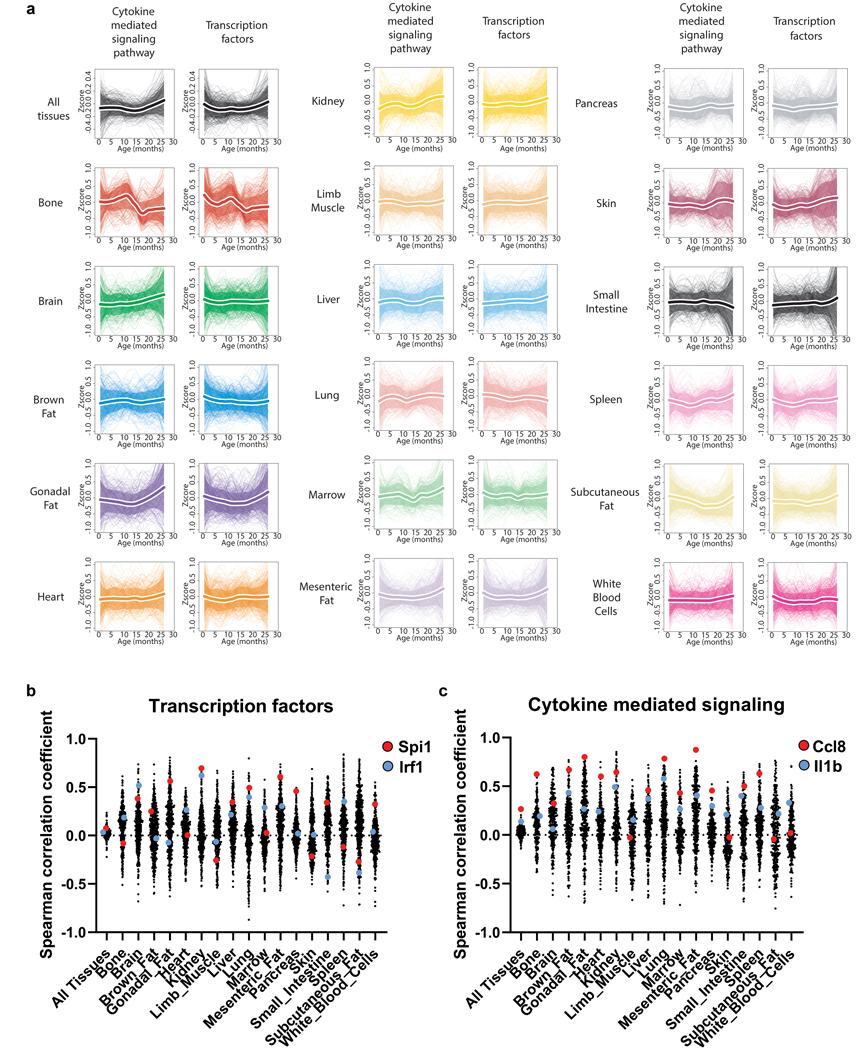
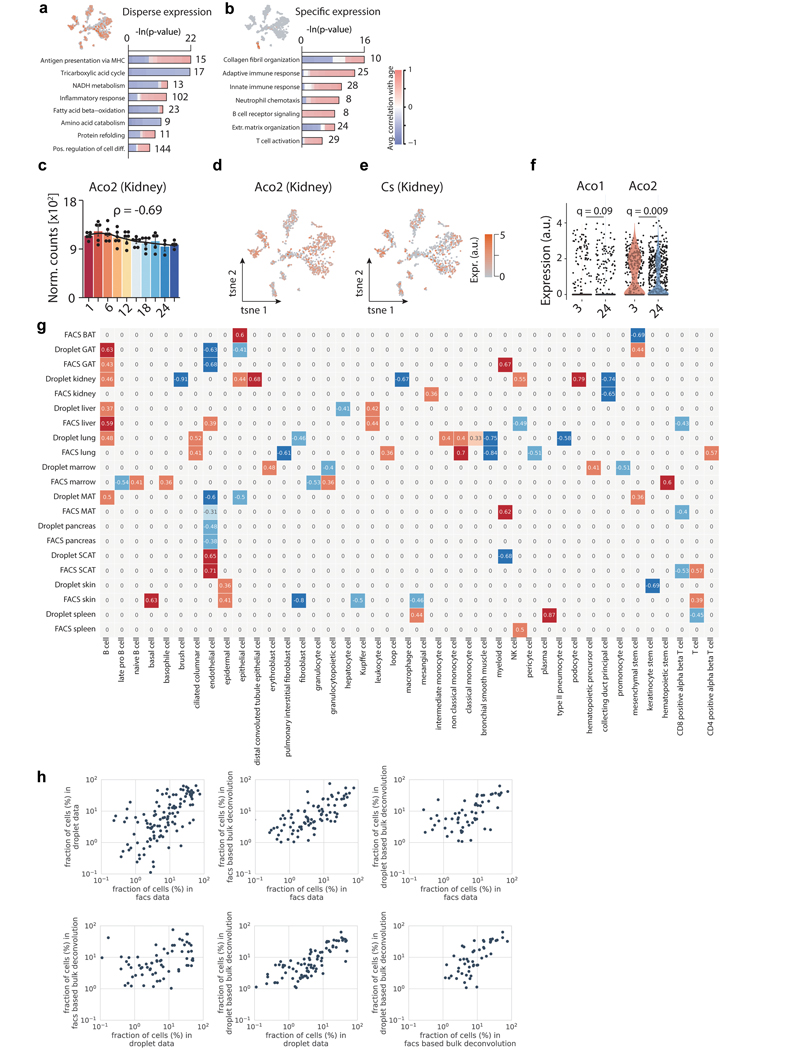
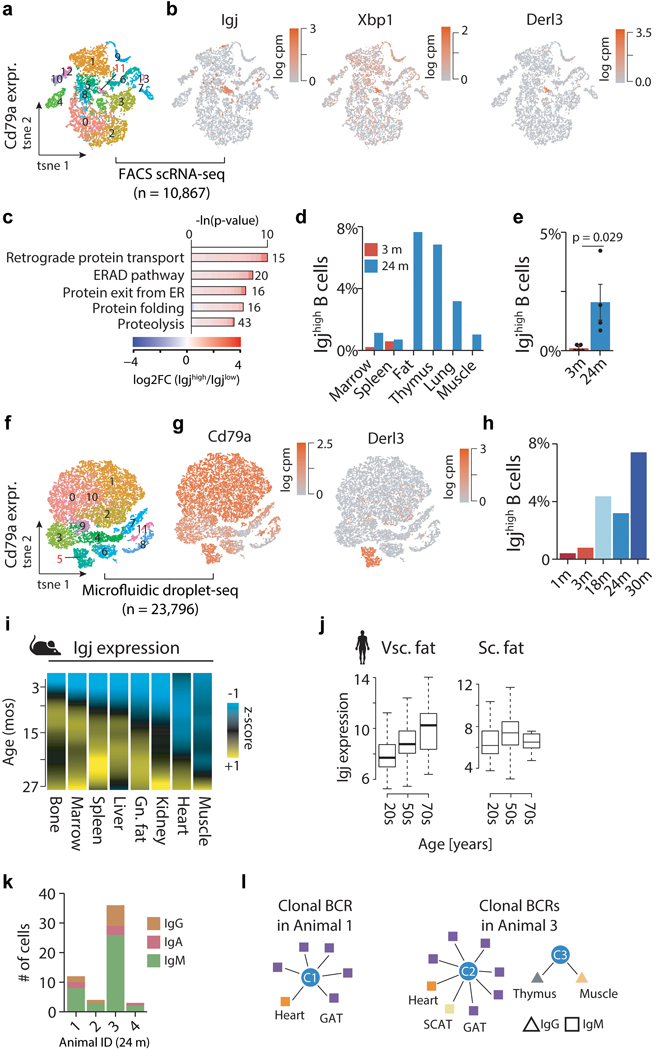
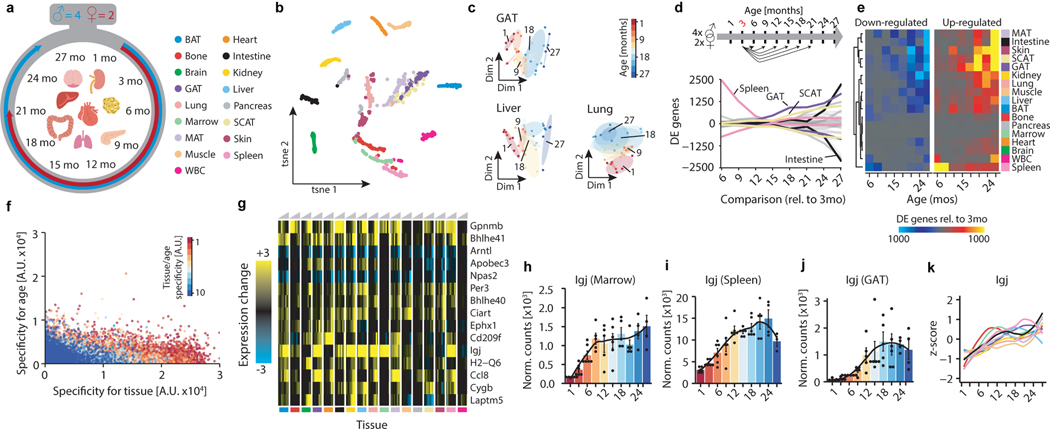
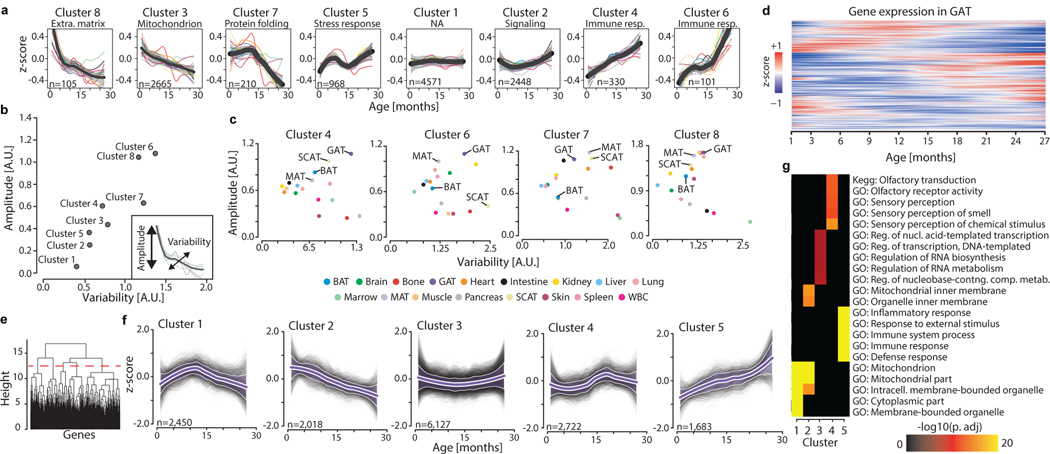
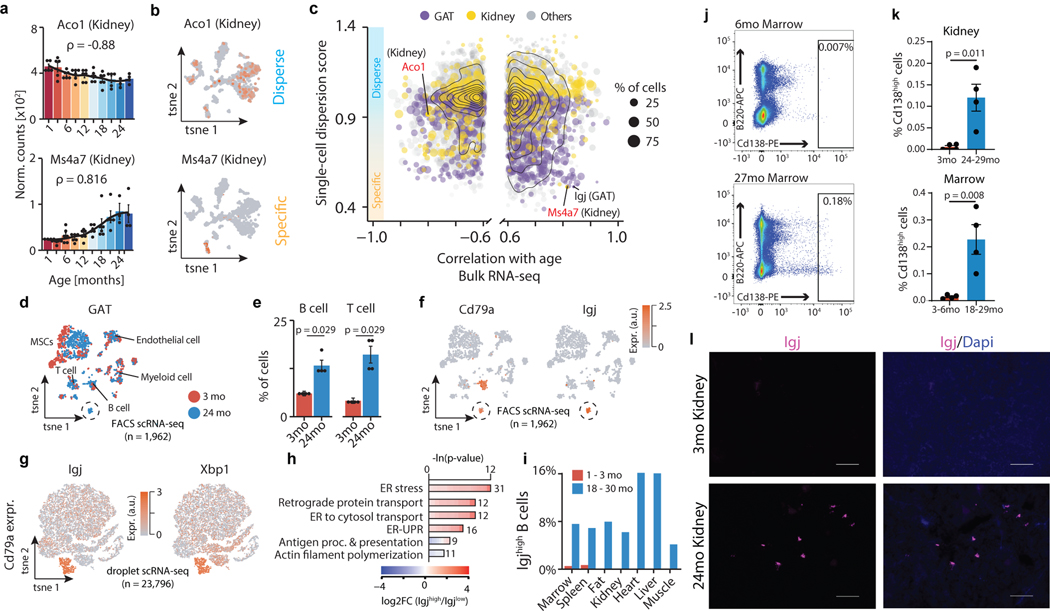
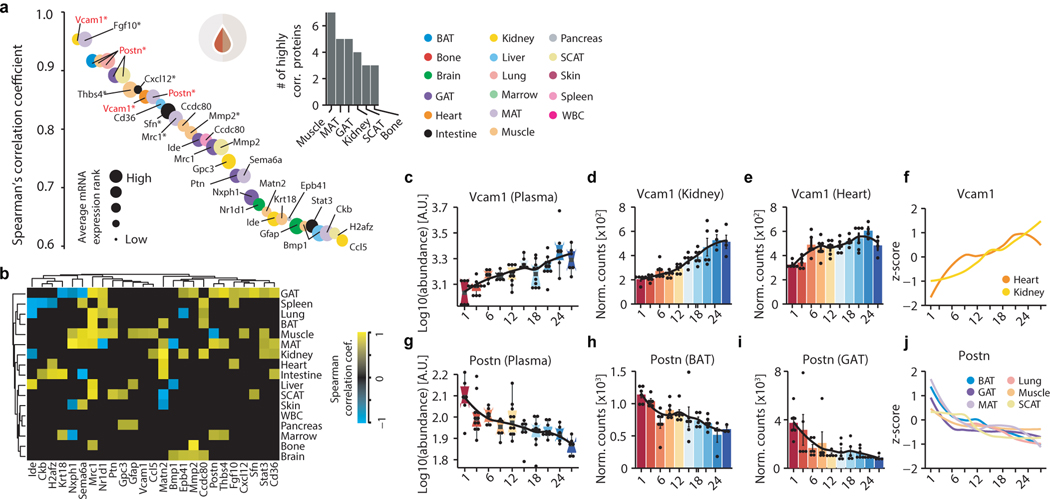
Ageing is the single greatest cause of disease and death worldwide, and understanding the associated processes could vastly improve quality of life. Although major categories of ageing damage have been identified-such as altered intercellular communication, loss of proteostasis and eroded mitochondrial function1-these deleterious processes interact with extraordinary complexity within and between organs, and a comprehensive, whole-organism analysis of ageing dynamics has been lacking. Here we performed bulk RNA sequencing of 17 organs and plasma proteomics at 10 ages across the lifespan of Mus musculus, and integrated these findings with data from the accompanying Tabula Muris Senis2-or 'Mouse Ageing Cell Atlas'-which follows on from the original Tabula Muris3. We reveal linear and nonlinear shifts in gene expression during ageing, with the associated genes clustered in consistent trajectory groups with coherent biological functions-including extracellular matrix regulation, unfolded protein binding, mitochondrial function, and inflammatory and immune response. Notably, these gene sets show similar expression across tissues, differing only in the amplitude and the age of onset of expression. Widespread activation of immune cells is especially pronounced, and is first detectable in white adipose depots during middle age. Single-cell RNA sequencing confirms the accumulation of T cells and B cells in adipose tissue-including plasma cells that express immunoglobulin J-which also accrue concurrently across diverse organs. Finally, we show how gene expression shifts in distinct tissues are highly correlated with corresponding protein levels in plasma, thus potentially contributing to the ageing of the systemic circulation. Together, these data demonstrate a similar yet asynchronous inter- and intra-organ progression of ageing, providing a foundation from which to track systemic sources of declining health at old age.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

References
-
- Kohonen T The self-organizing map. Proc. IEEE 78, 1464–1480 (1990).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
