

Single-cell lineage tracing unveils a role for TCF15 in haematopoiesis
- PMID: 32669716
- PMCID: PMC7579674
- DOI: 10.1038/s41586-020-2503-6
Single-cell lineage tracing unveils a role for TCF15 in haematopoiesis
Abstract
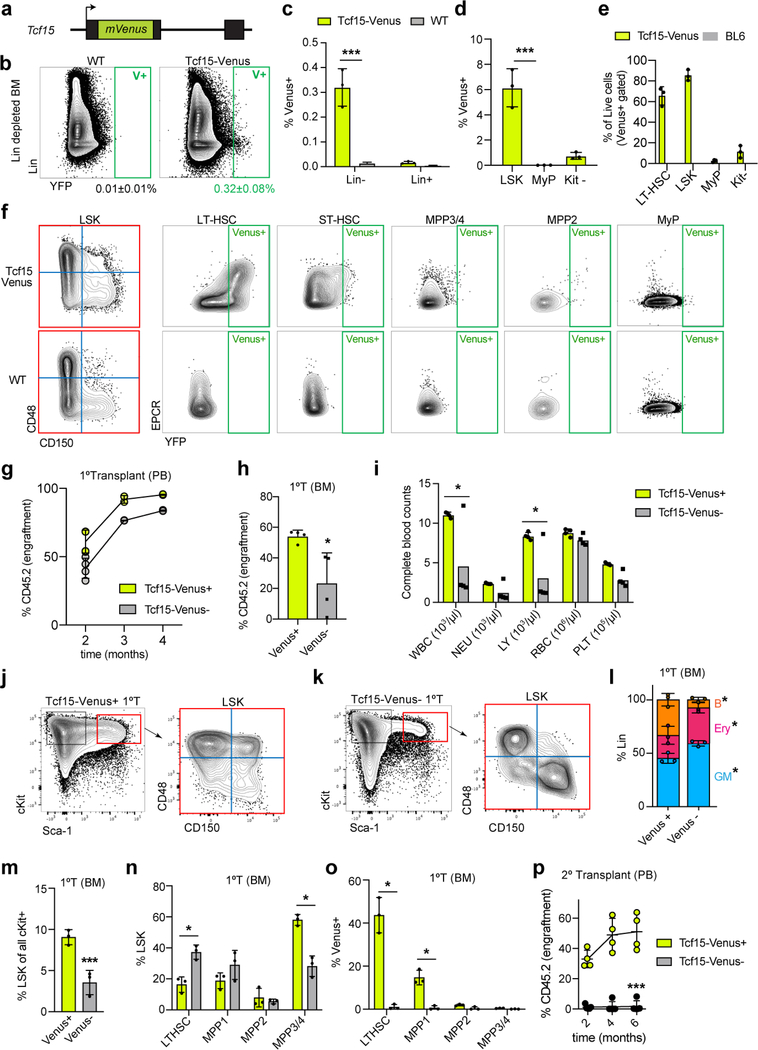
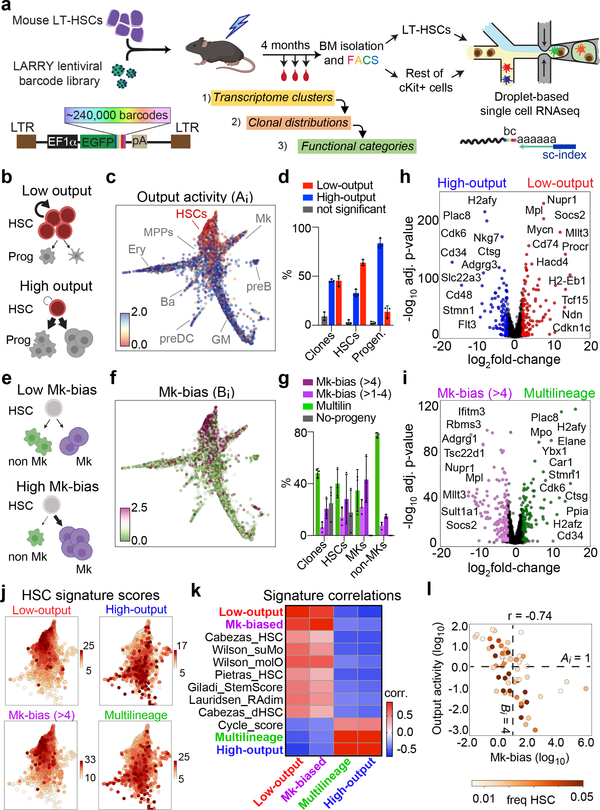
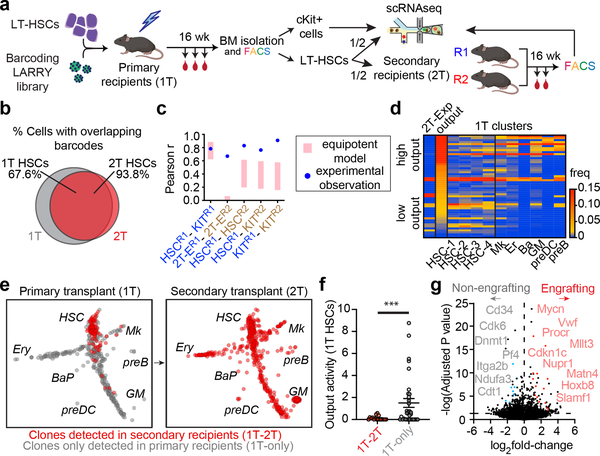
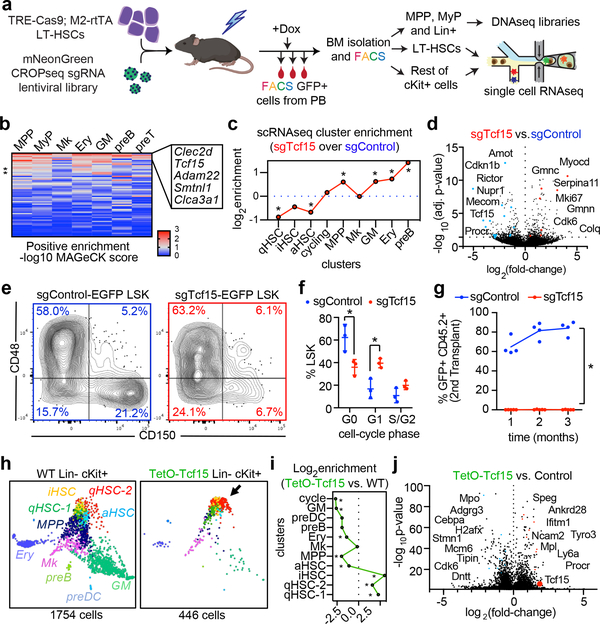
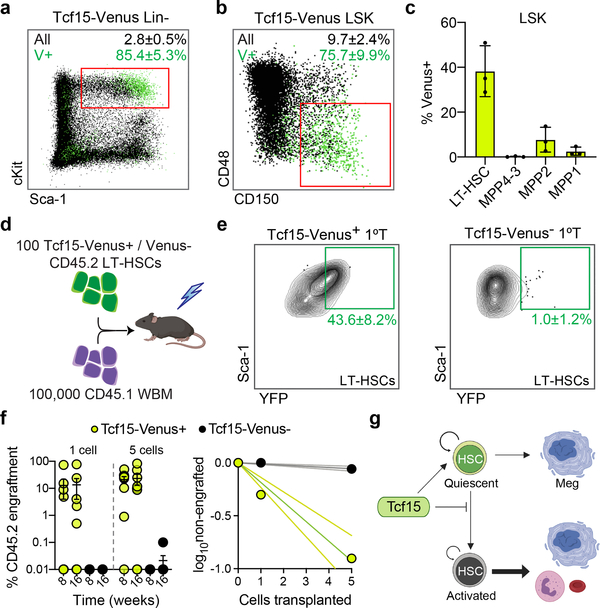
Bone marrow transplantation therapy relies on the life-long regenerative capacity of haematopoietic stem cells (HSCs)1,2. HSCs present a complex variety of regenerative behaviours at the clonal level, but the mechanisms underlying this diversity are still undetermined3-11. Recent advances in single-cell RNA sequencing have revealed transcriptional differences among HSCs, providing a possible explanation for their functional heterogeneity12-17. However, the destructive nature of sequencing assays prevents simultaneous observation of stem cell state and function. To solve this challenge, we implemented expressible lentiviral barcoding, which enabled simultaneous analysis of lineages and transcriptomes from single adult HSCs and their clonal trajectories during long-term bone marrow reconstitution. Analysis of differential gene expression between clones with distinct behaviour revealed an intrinsic molecular signature that characterizes functional long-term repopulating HSCs. Probing this signature through in vivo CRISPR screening, we found the transcription factor TCF15 to be required and sufficient to drive HSC quiescence and long-term self-renewal. In situ, Tcf15 expression labels the most primitive subset of true multipotent HSCs. In conclusion, our work elucidates clone-intrinsic molecular programmes associated with functional stem cell heterogeneity and identifies a mechanism for the maintenance of the self-renewing HSC state.
Conflict of interest statement
Competing interests
A.M.K. is a co-founder of 1cellbio, Ltd. The rest of the authors declare no competing interests.
Figures
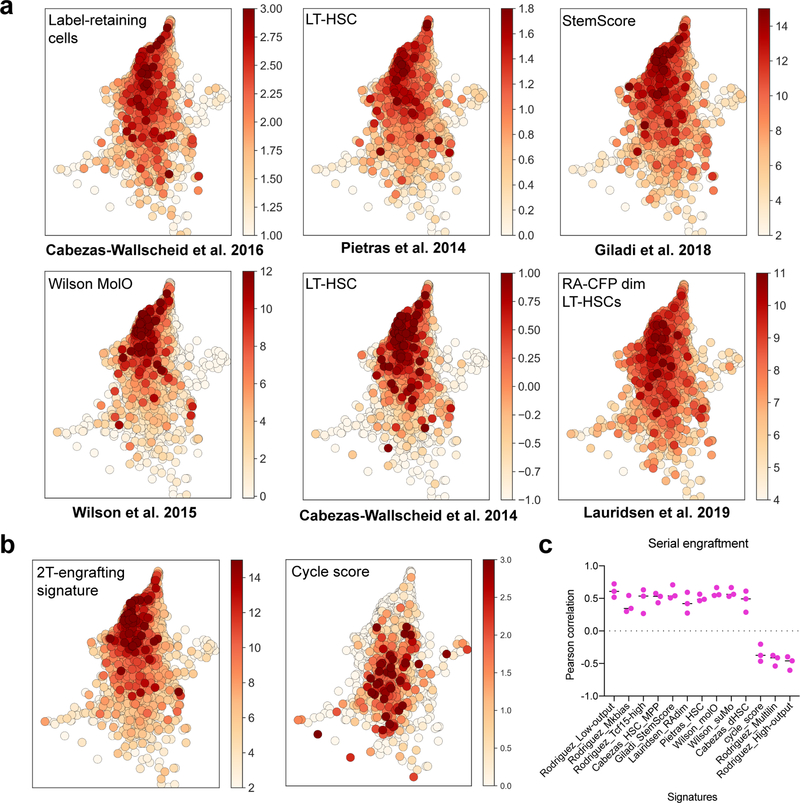
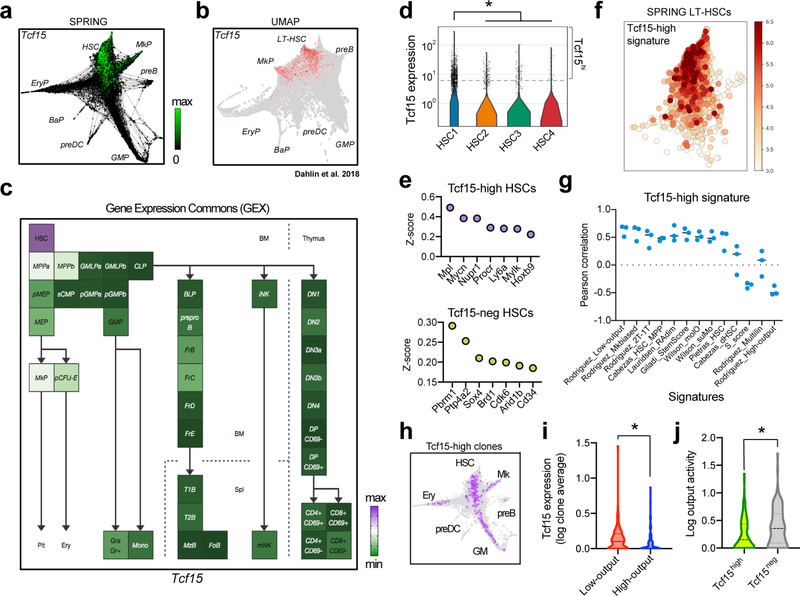
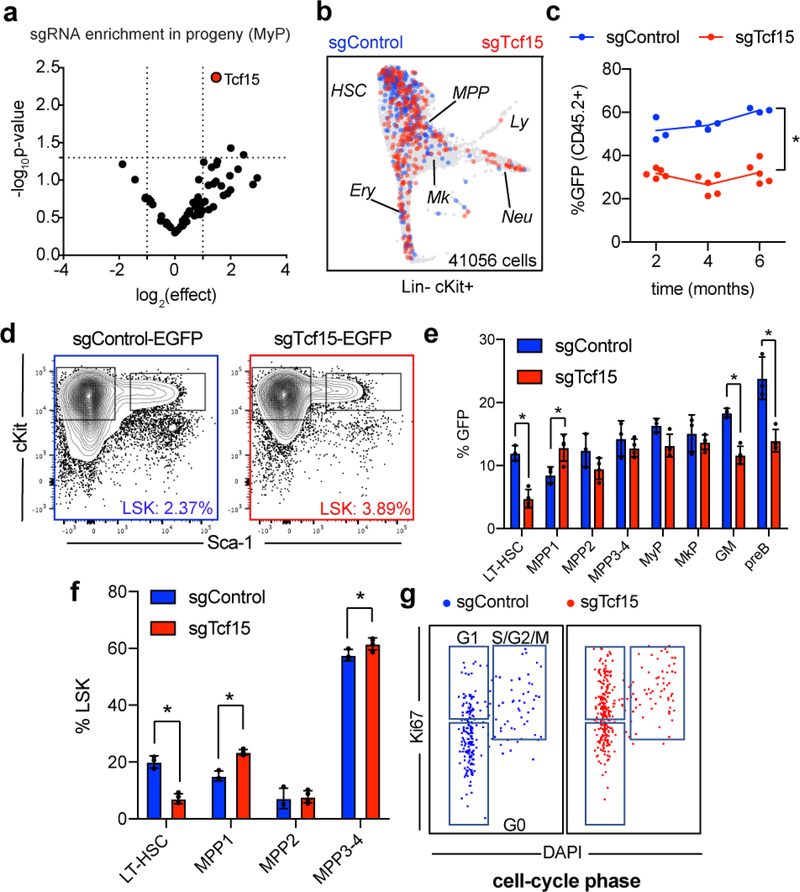
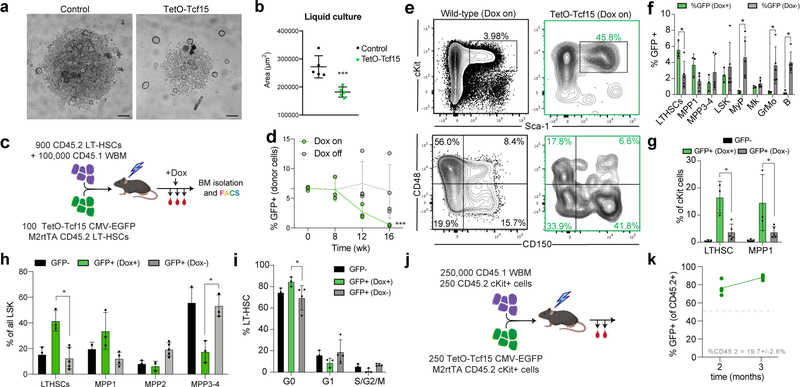
References
References (main-text)
-
- Haas S, Trumpp A & Milsom MD Causes and Consequences of Hematopoietic Stem Cell Heterogeneity. Cell Stem Cell 22, 627–638 (2018). - PubMed
-
- Naik SH et al. Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature 496, 229–232 (2013). - PubMed
-
- Dykstra B et al. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 1, 218–229 (2007). - PubMed
References (methods section)
-
- Zilionis R et al. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc 12, 44–73 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
