

Update in Laboratory Diagnosis of Thalassemia
- PMID: 32671092
- PMCID: PMC7326097
- DOI: 10.3389/fmolb.2020.00074
Update in Laboratory Diagnosis of Thalassemia
Abstract
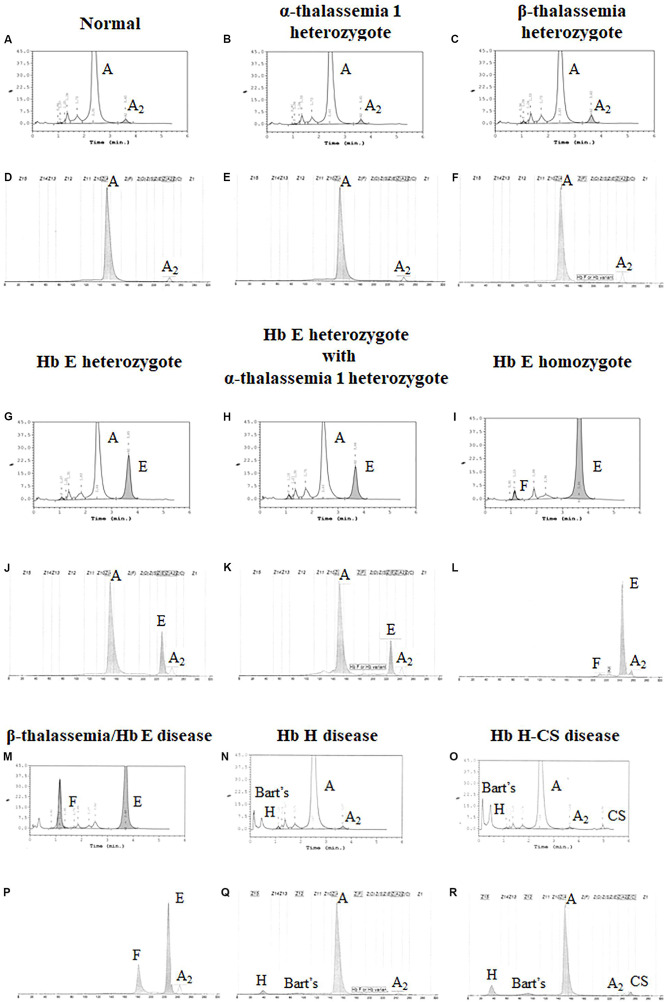
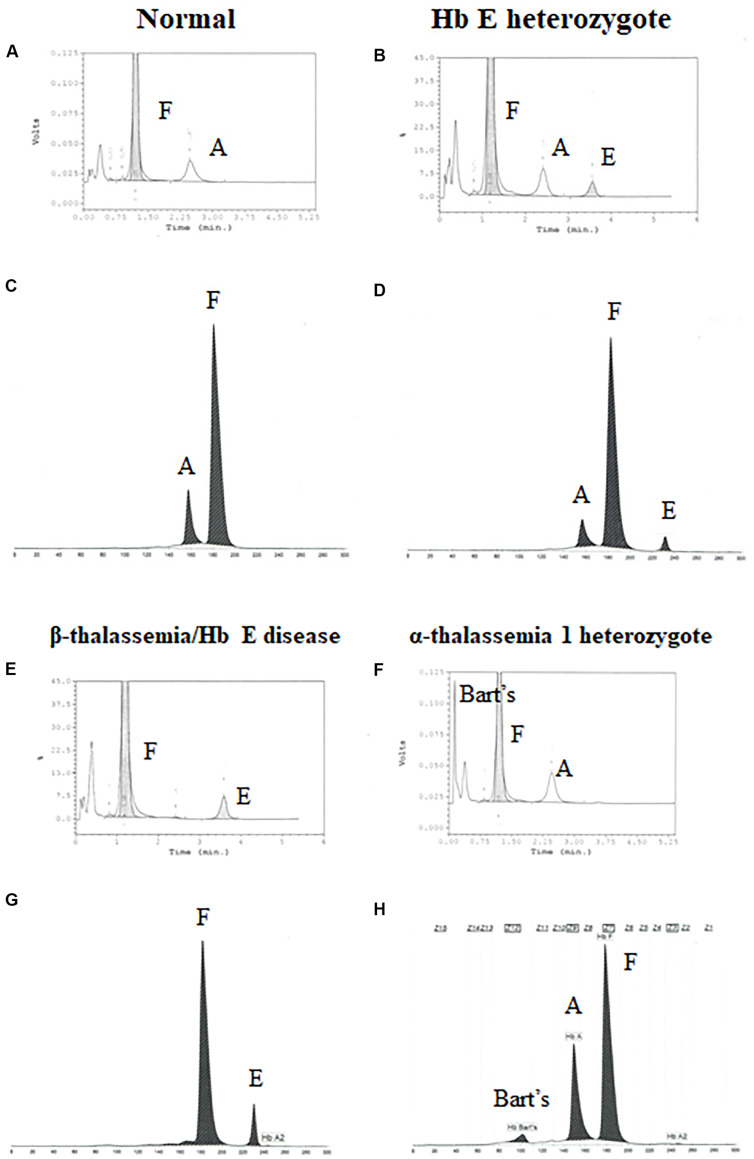
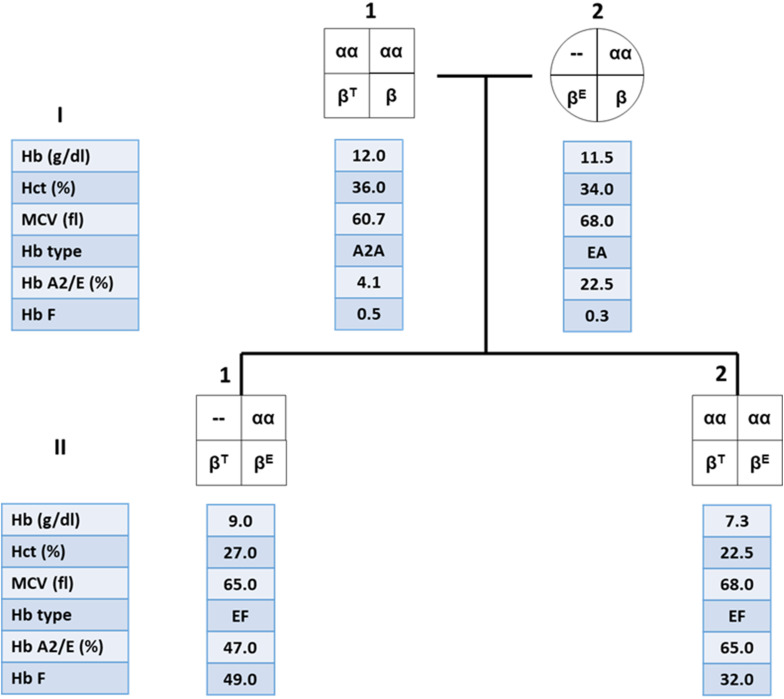
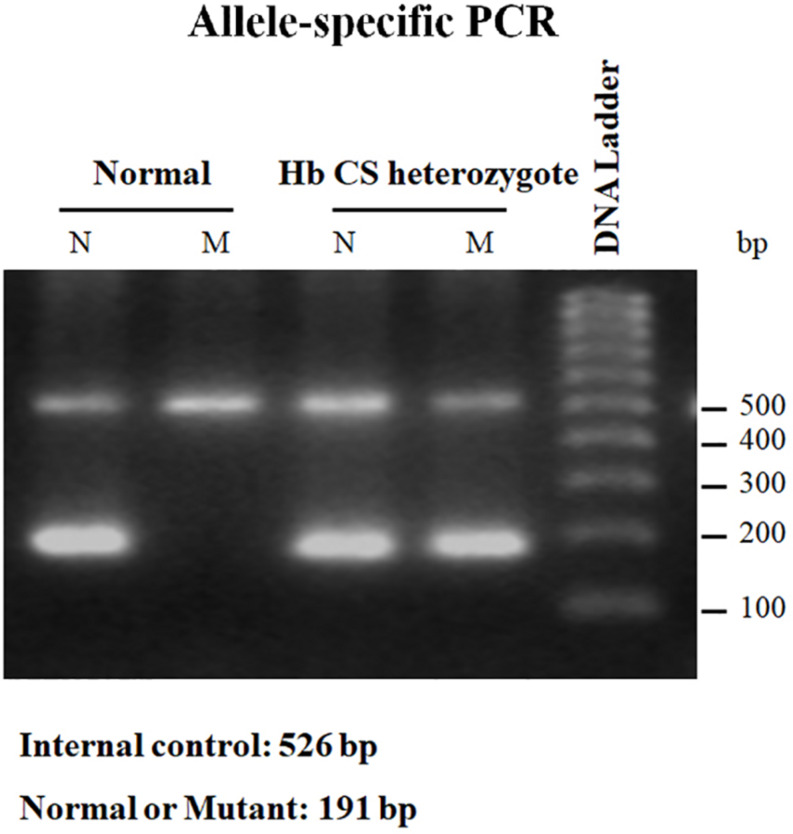
Alpha- and β-thalassemias and abnormal hemoglobin (Hb) are common in tropical countries. These abnormal globin genes in different combinations lead to many thalassemic diseases including three severe thalassemia diseases, i.e., homozygous β-thalassemia, β-thalassemia/Hb E, and Hb Bart's hydrops fetalis. Laboratory diagnosis of thalassemia requires a number of tests including red blood cell indices and Hb and DNA analyses. Thalassemic red blood cell analysis with an automated hematology analyzer is a primary screening for thalassemia since microcytosis and decreased Hb content of red blood cells are hallmarks of all thalassemic red blood cells. However, these two red blood cell indices cannot discriminate between thalassemia trait and iron deficiency or between α- and β-thalassemic conditions. Today, Hb analysis may be carried out by either automatic high-performance liquid chromatography (HPLC) or capillary zone electrophoresis (CE) system. These two systems give both qualitative and quantitative analysis of Hb components and help to do thalassemia prenatal and postnatal diagnoses within a short period. Both systems have a good correlation, but the interpretation under the CE system should be done with caution because Hb A2 is clearly separated from Hb E. In case of α-thalassemia gene interaction, it can affect the amount of Hb A2/E. Thalassemia genotypes can be characterized by the intensities between alpha-/beta-globin chains or alpha-/beta-mRNA ratios. However, those are presumptive diagnoses. Only DNA analysis can be made for specific thalassemia mutation diagnosis. Various molecular techniques have been used for point mutation detection in β-thalassemia and large-deletion detection in α-thalassemia. All of these techniques have some advantages and disadvantages. Recently, screening for both α- and β-thalassemia genes by next-generation sequencing (NGS) has been introduced. This technique gives an accurate diagnosis of thalassemia that may be misdiagnosed by other conventional techniques. The major limitation for using NGS in the screening of thalassemia is its cost which is still expensive. All service labs highly recommend to select the technique(s) they are most familiar and most economic one for their routine use.
Keywords: DNA analysis; diagnosis; hemoglobin analysis; hemoglobinopathies; thalassemia.
Copyright © 2020 Munkongdee, Chen, Winichagoon, Fucharoen and Paiboonsukwong.
Figures
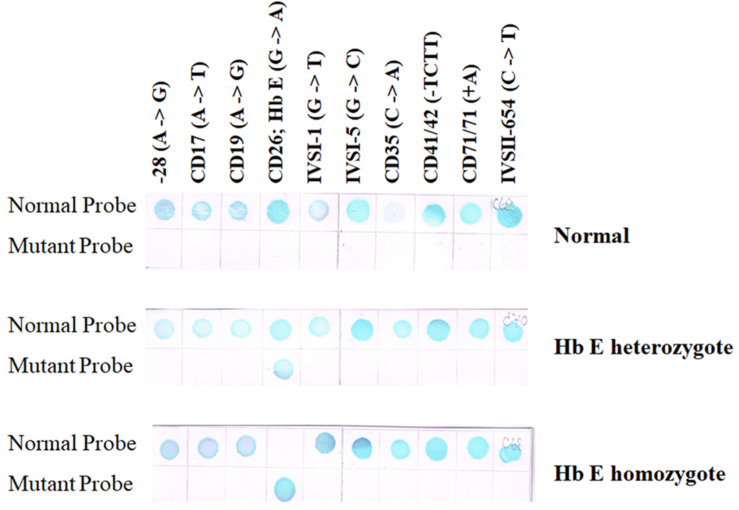
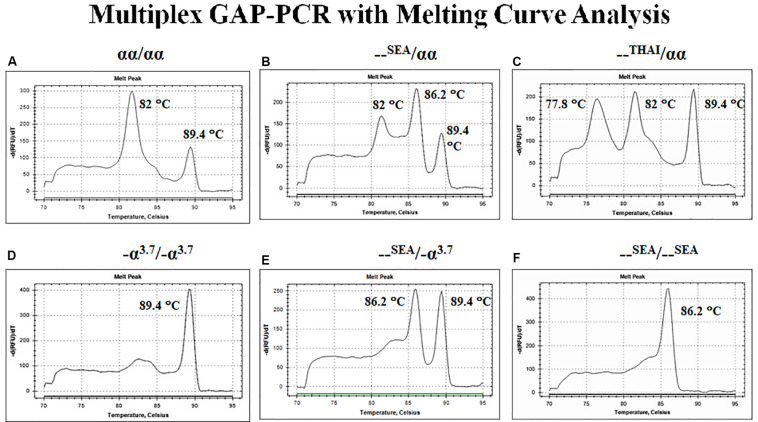
Similar articles
-
Laboratory Evaluation of Alpha Thalassemia.2024 Feb 9. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Feb 9. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 36508547 Free Books & Documents.
-
Prenatal and postnatal diagnoses of thalassemias and hemoglobinopathies by HPLC.Clin Chem. 1998 Apr;44(4):740-8. Clin Chem. 1998. PMID: 9554484
-
Prenatal diagnosis of thalassemia and hemoglobinopathies in Thailand: experience from 100 pregnancies.Southeast Asian J Trop Med Public Health. 1991 Mar;22(1):16-29. Southeast Asian J Trop Med Public Health. 1991. PMID: 1948258 Review.
-
Thalassemia and abnormal hemoglobin.Int J Hematol. 2002 Aug;76 Suppl 2:83-9. doi: 10.1007/BF03165094. Int J Hematol. 2002. PMID: 12430905 Review.
-
Fetal red blood cell parameters in thalassemia and hemoglobinopathies.Fetal Diagn Ther. 2013;34(3):166-71. doi: 10.1159/000354343. Epub 2013 Sep 14. Fetal Diagn Ther. 2013. PMID: 24051385
Cited by
-
Analysis of genotype-phenotype correlation in patients with α-thalassemia from Fujian province, Southeastern China.J Clin Lab Anal. 2022 Oct;36(10):e24696. doi: 10.1002/jcla.24696. Epub 2022 Sep 13. J Clin Lab Anal. 2022. PMID: 36099017 Free PMC article.
-
Thalassemia: A Review of the Challenges to the Families and Caregivers.Cureus. 2022 Dec 13;14(12):e32491. doi: 10.7759/cureus.32491. eCollection 2022 Dec. Cureus. 2022. PMID: 36523854 Free PMC article. Review.
-
α- and β-Genotyping of Thalassemia Patients Based on a Multimodal Liver MRI Radiomics Model: A Preliminary Study in Two Centers.Diagnostics (Basel). 2023 Mar 3;13(5):958. doi: 10.3390/diagnostics13050958. Diagnostics (Basel). 2023. PMID: 36900102 Free PMC article.
-
Molecular analysis of alpha- and beta-thalassemia in Meizhou region and comparison of gene mutation spectrum with different regions of southern China.J Clin Lab Anal. 2021 Dec;35(12):e24105. doi: 10.1002/jcla.24105. Epub 2021 Nov 9. J Clin Lab Anal. 2021. PMID: 34752669 Free PMC article.
-
A next-generation sequencing-based universal target panel and algorithm for one-stop detection of copy number alterations and single-nucleotide variations in the HBB gene cluster for rapid diagnosis of β-thalassemia.Mol Biol Rep. 2025 Jan 16;52(1):128. doi: 10.1007/s11033-024-10196-2. Mol Biol Rep. 2025. PMID: 39820710
References
-
- Barbara J. B. (2006). “The α, β, (and (thalassemias and related conditions,” in Haemoglobinopathy Diagnosis, ed. Barbara J. B. (London: Blackwell; ), 63–138.
-
- Calzolari R., McMorrow T., Yannoutsos N., Langeveld A., Grosveld F. (1999). Deletion of a region that is a candidate for the difference between the deletion forms of hereditary persistence of fetal hemoglobin and deltabeta-thalassemia affects beta- but not gamma-globin gene expression. EMBO J. 18 949–958. 10.1093/emboj/18.4.949 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources