

Severe Autoinflammatory Manifestations and Antibody Deficiency Due to Novel Hypermorphic PLCG2 Mutations
- PMID: 32671674
- PMCID: PMC7505877
- DOI: 10.1007/s10875-020-00794-7
Severe Autoinflammatory Manifestations and Antibody Deficiency Due to Novel Hypermorphic PLCG2 Mutations
Abstract
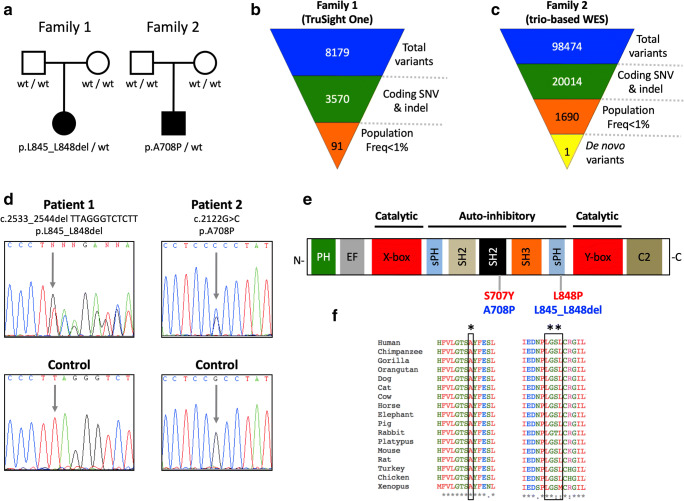
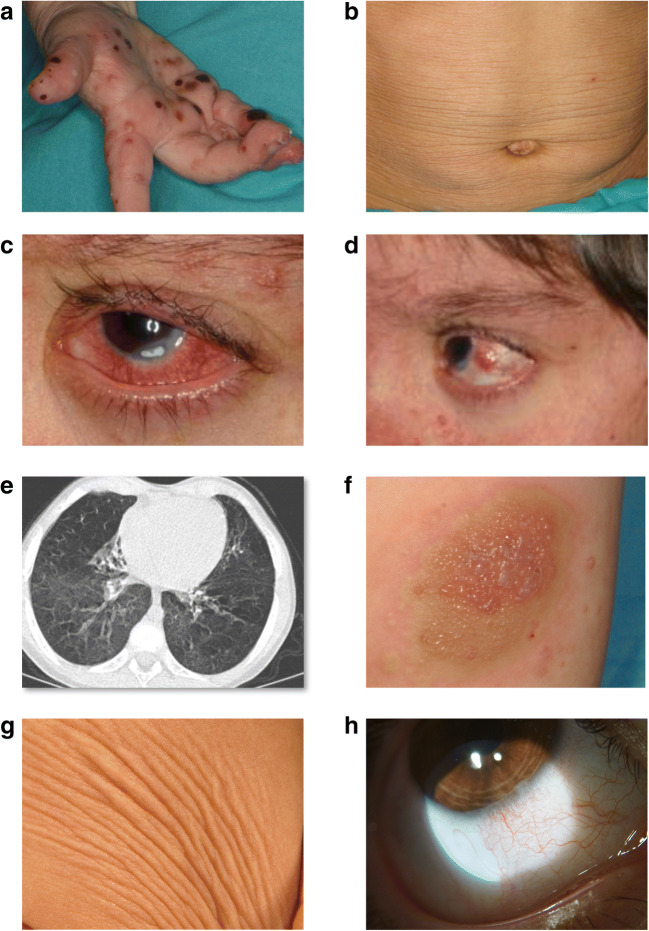
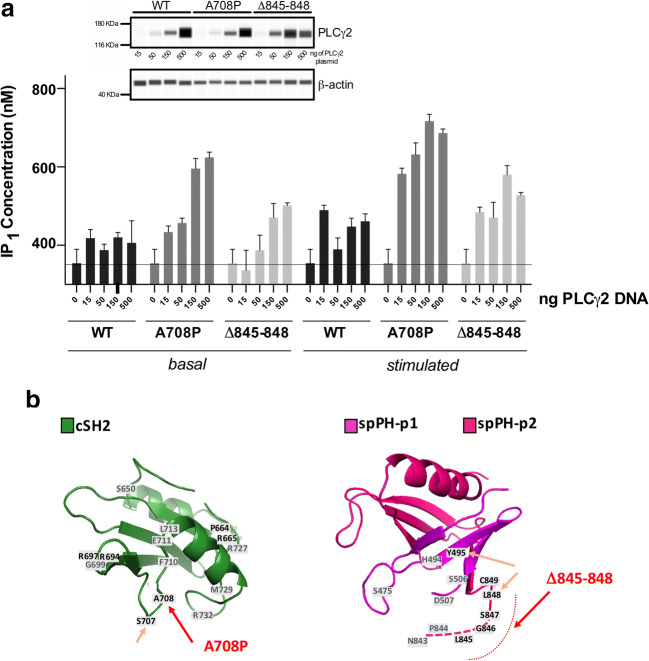
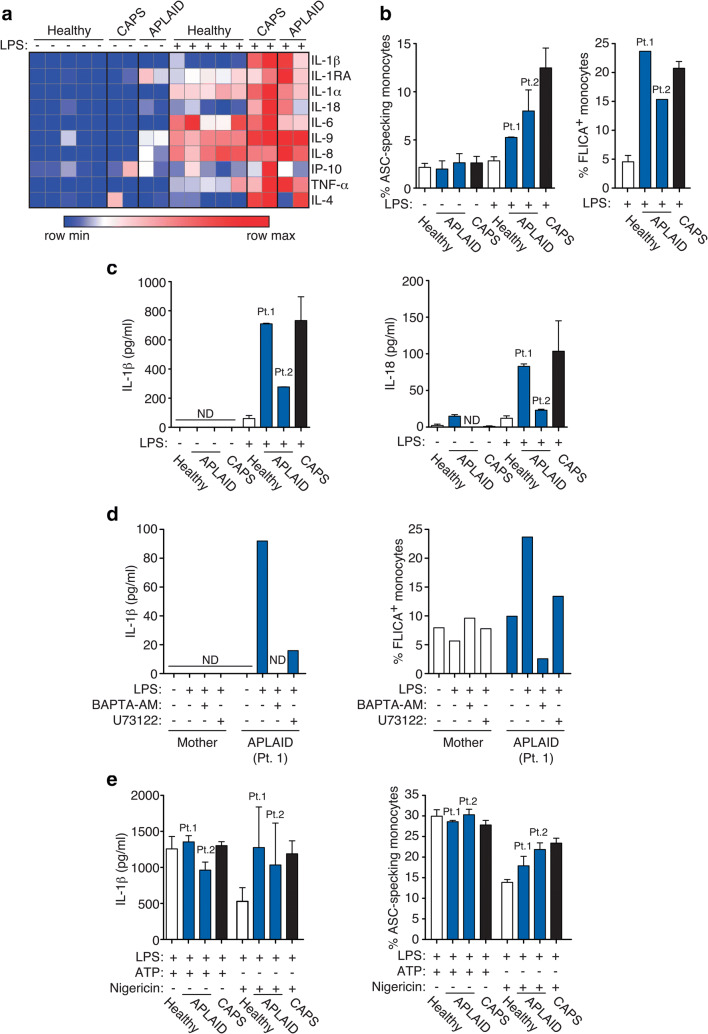
Autoinflammatory diseases (AIDs) were first described as clinical disorders characterized by recurrent episodes of seemingly unprovoked sterile inflammation. In the past few years, the identification of novel AIDs expanded their phenotypes toward more complex clinical pictures associating vasculopathy, autoimmunity, or immunodeficiency. Herein, we describe two unrelated patients suffering since the neonatal period from a complex disease mainly characterized by severe sterile inflammation, recurrent bacterial infections, and marked humoral immunodeficiency. Whole-exome sequencing detected a novel, de novo heterozygous PLCG2 variant in each patient (p.Ala708Pro and p.Leu845_Leu848del). A clear enhanced PLCγ2 activity for both variants was demonstrated by both ex vivo calcium responses of the patient's B cells to IgM stimulation and in vitro assessment of PLC activity. These data supported the autoinflammation and PLCγ2-associated antibody deficiency and immune dysregulation (APLAID) diagnosis in both patients. Immunological evaluation revealed a severe decrease of immunoglobulins and B cells, especially class-switched memory B cells, with normal T and NK cell counts. Analysis of bone marrow of one patient revealed a reduced immature B cell fraction compared with controls. Additional investigations showed that both PLCG2 variants activate the NLRP3-inflammasome through the alternative pathway instead of the canonical pathway. Collectively, the evidences here shown expand APLAID diversity toward more severe phenotypes than previously reported including dominantly inherited agammaglobulinemia, add novel data about its genetic basis, and implicate the alternative NLRP3-inflammasome activation pathway in the basis of sterile inflammation.
Keywords: APLAID; Autoinflammatory diseases; PLCγ2; agammaglobulinemia; caspase-1; inflammasome; interleukin-1.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, Lee CCR, DiMattia MA, Cowen EW, Gonzalez B, Palmer I, DiGiovanna JJ, Biancotto A, Kim H, Tsai WL, Trier AM, Huang Y, Stone DL, Hill S, Kim HJ, St. Hilaire C, Gurprasad S, Plass N, Chapelle D, Horkayne-Szakaly I, Foell D, Barysenka A, Candotti F, Holland SM, Hughes JD, Mehmet H, Issekutz AC, Raffeld M, McElwee J, Fontana JR, Minniti CP, Moir S, Kastner DL, Gadina M, Steven AC, Wingfield PT, Brooks SR, Rosenzweig SD, Fleisher TA, Deng Z, Boehm M, Paller AS, Goldbach-Mansky R. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. - DOI - PMC - PubMed
-
- Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, Stone DL, Chae JJ, Rosenzweig SD, Bishop K, Barron KS, Kuehn HS, Hoffmann P, Negro A, Tsai WL, Cowen EW, Pei W, Milner JD, Silvin C, Heller T, Chin DT, Patronas NJ, Barber JS, Lee CCR, Wood GM, Ling A, Kelly SJ, Kleiner DE, Mullikin JC, Ganson NJ, Kong HH, Hambleton S, Candotti F, Quezado MM, Calvo KR, Alao H, Barham BK, Jones A, Meschia JF, Worrall BB, Kasner SE, Rich SS, Goldbach-Mansky R, Abinun M, Chalom E, Gotte AC, Punaro M, Pascual V, Verbsky JW, Torgerson TR, Singer NG, Gershon TR, Ozen S, Karadag O, Fleisher TA, Remmers EF, Burgess SM, Moir SL, Gadina M, Sood R, Hershfield MS, Boehm M, Kastner DL, Aksentijevich I. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370:911–920. doi: 10.1056/NEJMoa1307361. - DOI - PMC - PubMed
-
- Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, Zlotogorski A, Berkun Y, Press JJ, Mukamel M, Voth I, Hashkes PJ, Harel L, Hoffer V, Ling E, Yalcinkaya F, Kasapcopur O, Lee MK, Klevit RE, Renbaum P, Weinberg-Shukron A, Sener EF, Schormair B, Zeligson S, Marek-Yagel D, Strom TM, Shohat M, Singer A, Rubinow A, Pras E, Winkelmann J, Tekin M, Anikster Y, King MC, Levy-Lahad E. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014;370:921–931. doi: 10.1056/NEJMoa1307362. - DOI - PubMed
-
- Chakraborty PK, Schmitz-Abe K, Kennedy EK, Mamady H, Naas T, Durie D, Campagna DR, Lau A, Sendamarai AK, Wiseman DH, May A, Jolles S, Connor P, Powell C, Heeney MM, Giardina PJ, Klaassen RJ, Kannengiesser C, Thuret I, Thompson AA, Marques L, Hughes S, Bonney DK, Bottomley SS, Wynn RF, Laxer RM, Minniti CP, Moppett J, Bordon V, Geraghty M, Joyce PBM, Markianos K, Rudner AD, Holcik M, Fleming MD. Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD) Blood. 2014;124:2867–2871. doi: 10.1182/blood-2014-08-591370. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
