

Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation
- PMID: 32673537
- PMCID: PMC7982967
- DOI: 10.1161/CIRCRESAHA.120.316958
Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation
Abstract
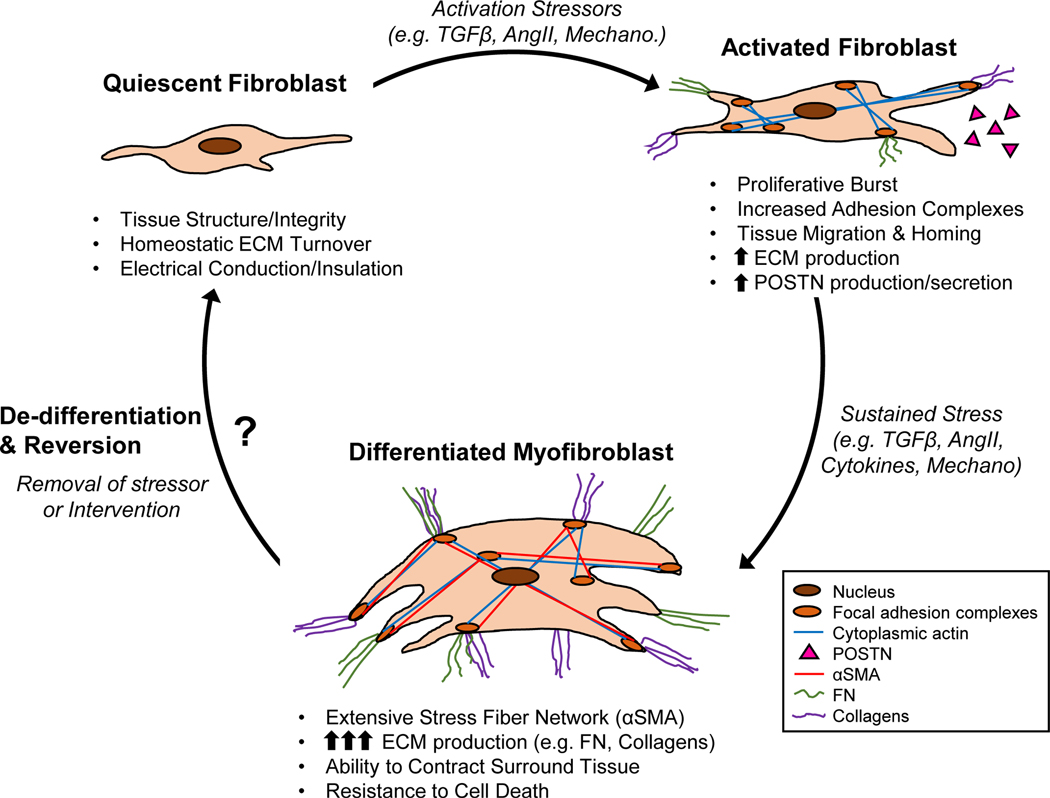
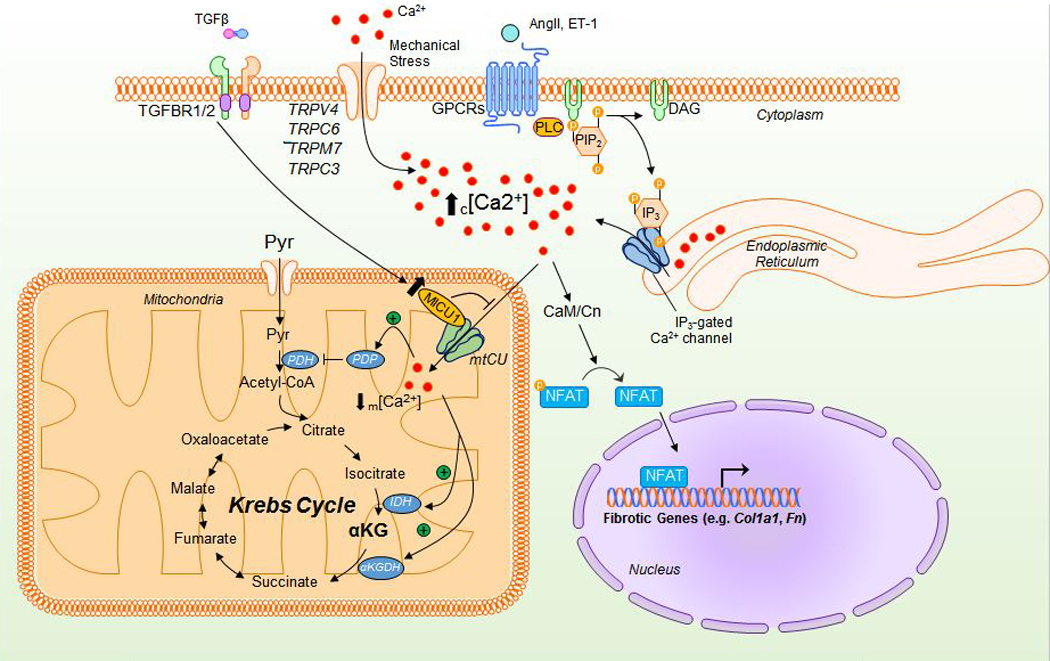
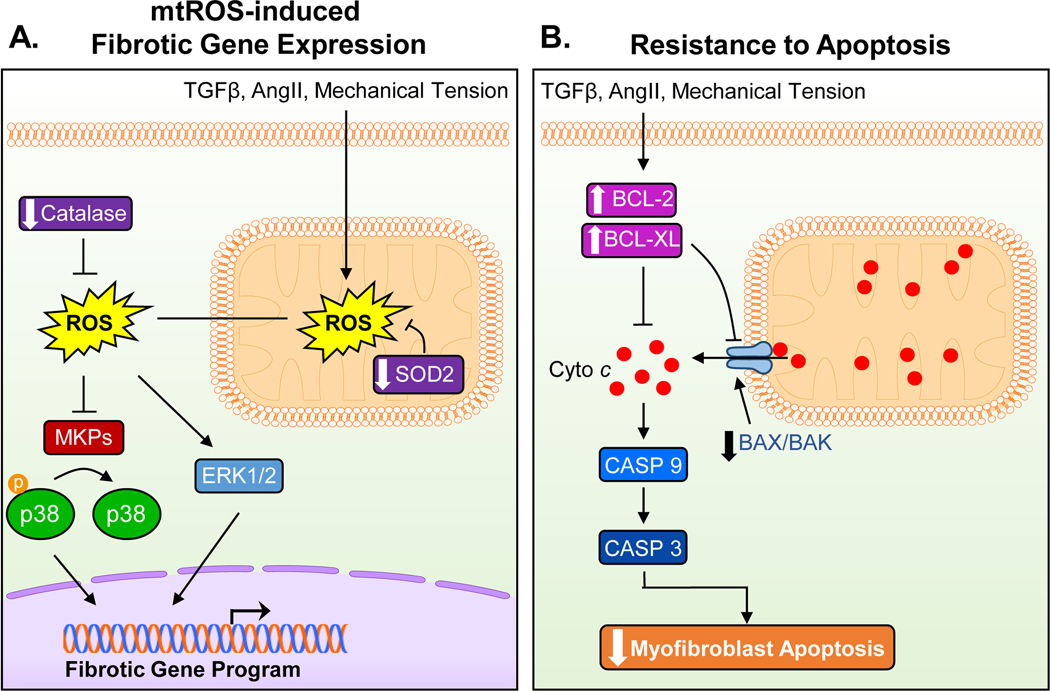
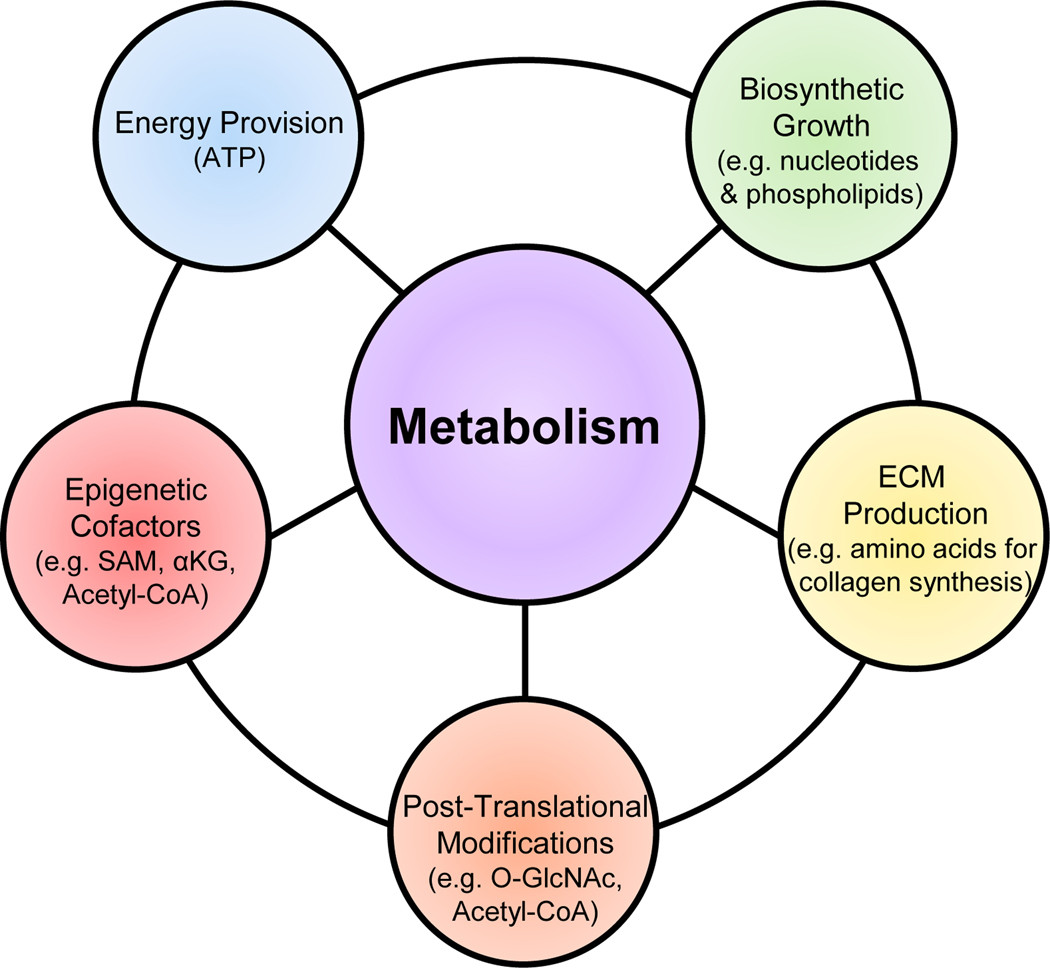
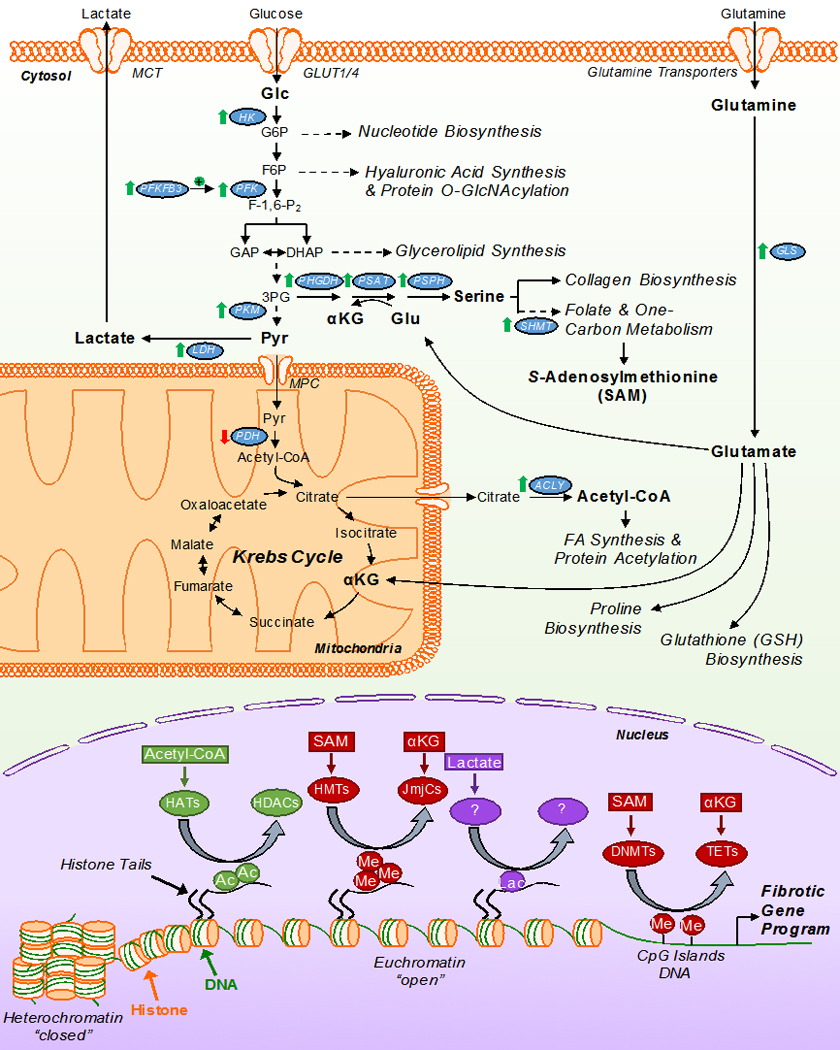
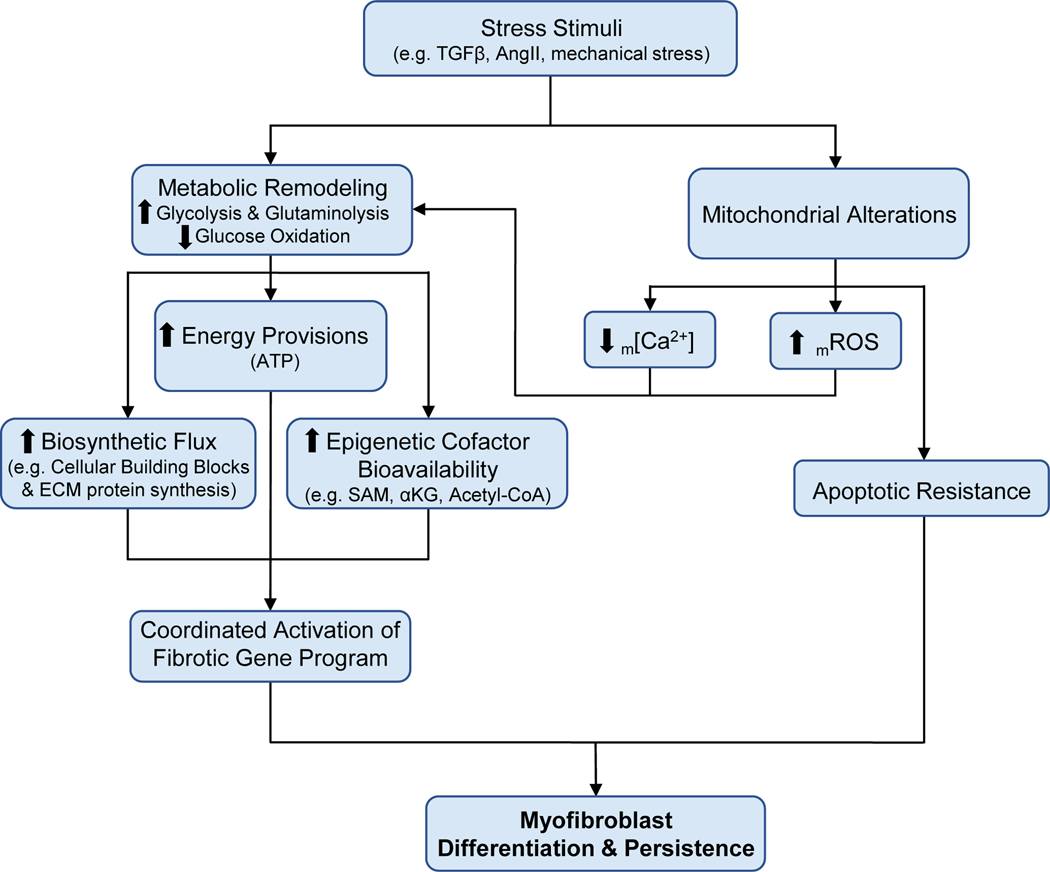
Cardiac fibrosis is mediated by the activation of resident cardiac fibroblasts, which differentiate into myofibroblasts in response to injury or stress. Although myofibroblast formation is a physiological response to acute injury, such as myocardial infarction, myofibroblast persistence, as occurs in heart failure, contributes to maladaptive remodeling and progressive functional decline. Although traditional pathways of activation, such as TGFβ (transforming growth factor β) and AngII (angiotensin II), have been well characterized, less understood are the alterations in mitochondrial function and cellular metabolism that are necessary to initiate and sustain myofibroblast formation and function. In this review, we highlight recent reports detailing the mitochondrial and metabolic mechanisms that contribute to myofibroblast differentiation, persistence, and function with the hope of identifying novel therapeutic targets to treat, and potentially reverse, tissue organ fibrosis.
Keywords: fibrosis; heart failure; metabolism; mitochondria; myofibroblast.
Figures
References
-
- Michel G, Tonon T, Scornet D, Cock JM and Kloareg B. The cell wall polysaccharide metabolism of the brown alga Ectocarpus siliculosus. Insights into the evolution of extracellular matrix polysaccharides in Eukaryotes. New Phytol. 2010;188:82–97. - PubMed
-
- Theocharis AD, Skandalis SS, Gialeli C and Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev. 2016;97:4–27. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
