

Inflammation in Parkinson's Disease: Mechanisms and Therapeutic Implications
- PMID: 32674367
- PMCID: PMC7408280
- DOI: 10.3390/cells9071687
Inflammation in Parkinson's Disease: Mechanisms and Therapeutic Implications
Abstract
Parkinson's disease (PD) is a common neurodegenerative disorder primarily characterized by the death of dopaminergic neurons that project from the substantia nigra pars compacta. Although the molecular bases for PD development are still little defined, extensive evidence from human samples and animal models support the involvement of inflammation in onset or progression. However, the exact trigger for this response remains unclear. Here, we provide a systematic review of the cellular mediators, i.e., microglia, astroglia and endothelial cells. We also discuss the genetic and transcriptional control of inflammation in PD and the immunomodulatory role of dopamine and reactive oxygen species. Finally, we summarize the preclinical and clinical approaches targeting neuroinflammation in PD.
Keywords: Parkinson’s disease; immune system; neurodegeneration; neuroinflammation; therapy.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
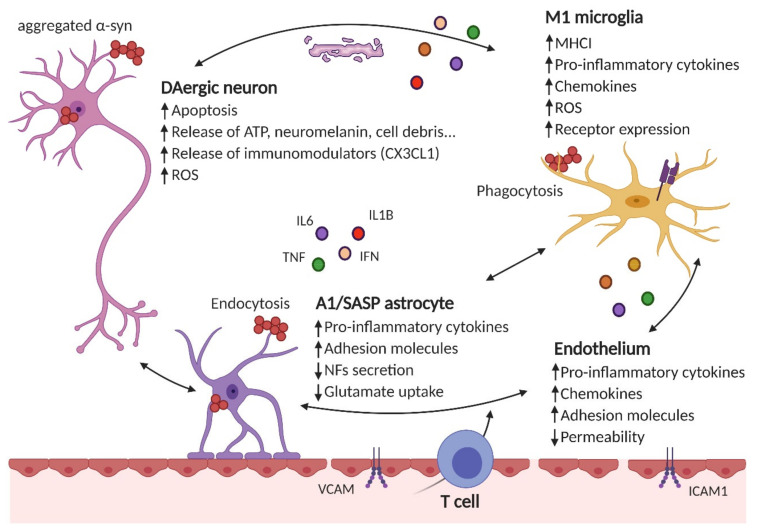
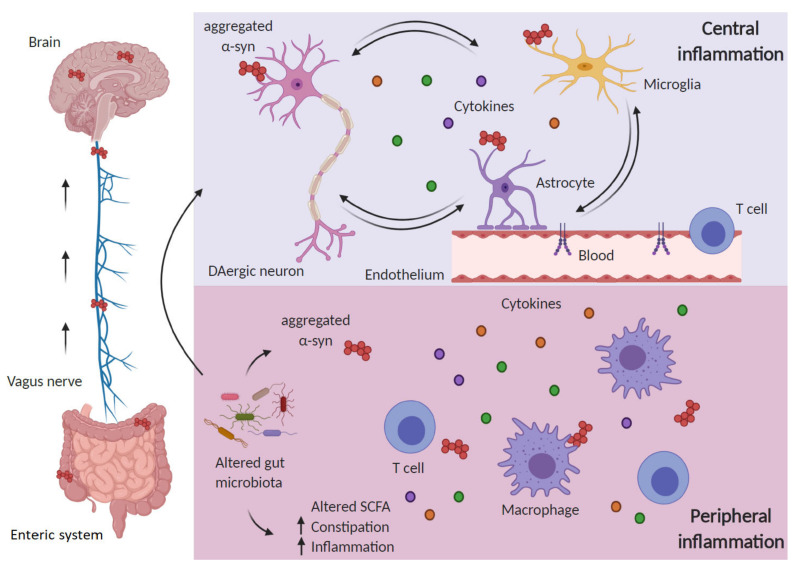
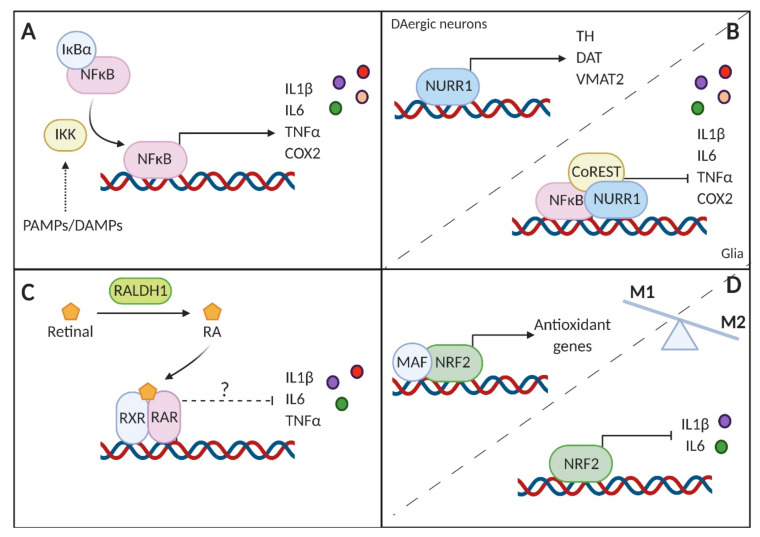
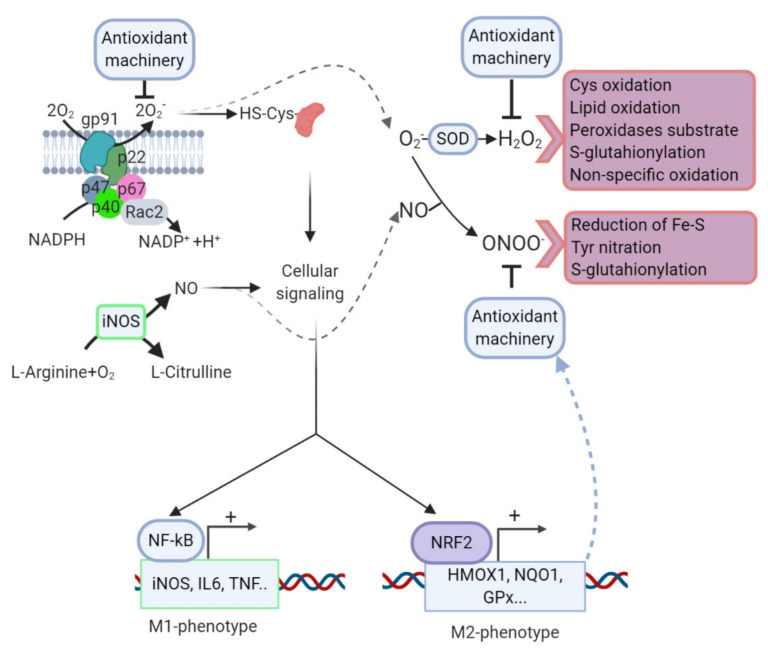
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
