

Role of NAD+-Modulated Mitochondrial Free Radical Generation in Mechanisms of Acute Brain Injury
- PMID: 32674501
- PMCID: PMC7408119
- DOI: 10.3390/brainsci10070449
Role of NAD+-Modulated Mitochondrial Free Radical Generation in Mechanisms of Acute Brain Injury
Abstract
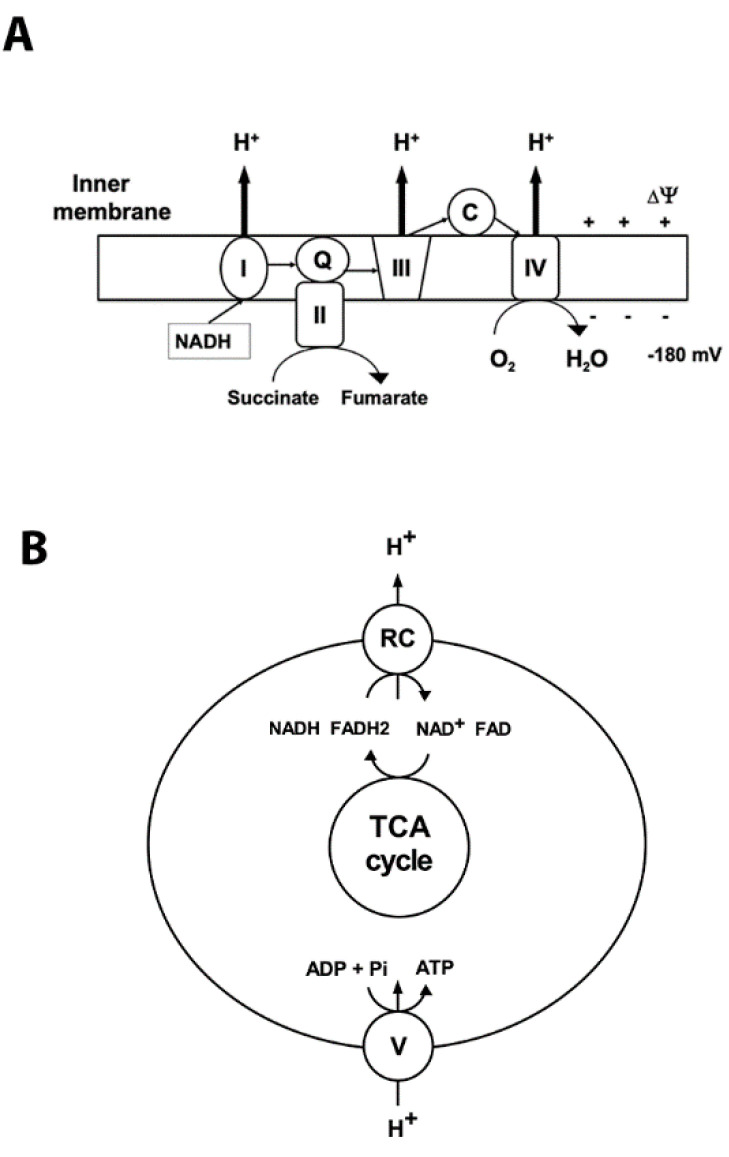
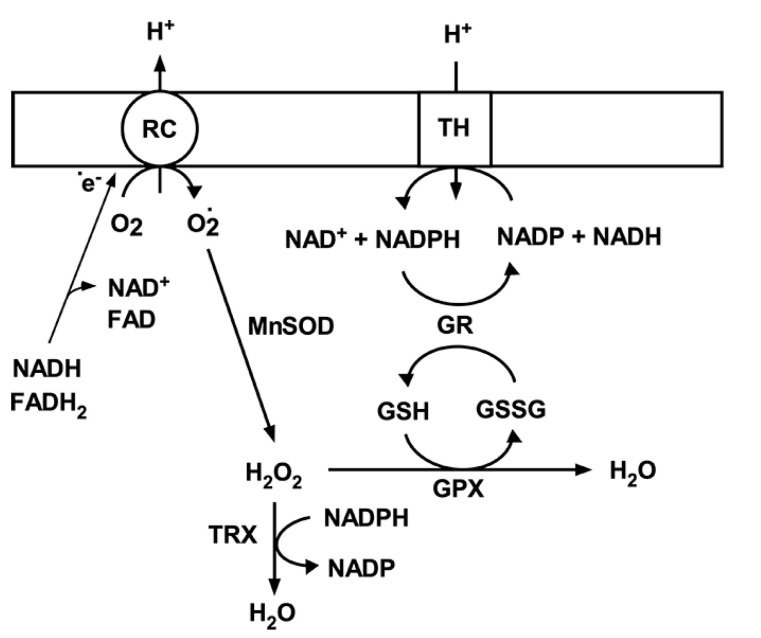
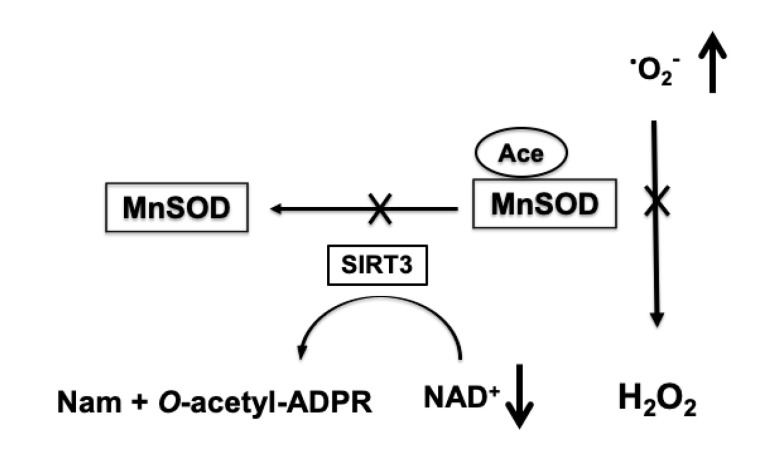
It is commonly accepted that mitochondria represent a major source of free radicals following acute brain injury or during the progression of neurodegenerative diseases. The levels of reactive oxygen species (ROS) in cells are determined by two opposing mechanisms-the one that produces free radicals and the cellular antioxidant system that eliminates ROS. Thus, the balance between the rate of ROS production and the efficiency of the cellular detoxification process determines the levels of harmful reactive oxygen species. Consequently, increase in free radical levels can be a result of higher rates of ROS production or due to the inhibition of the enzymes that participate in the antioxidant mechanisms. The enzymes' activity can be modulated by post-translational modifications that are commonly altered under pathologic conditions. In this review we will discuss the mechanisms of mitochondrial free radical production following ischemic insult, mechanisms that protect mitochondria against free radical damage, and the impact of post-ischemic nicotinamide adenine mononucleotide (NAD+) catabolism on mitochondrial protein acetylation that affects ROS generation and mitochondrial dynamics. We propose a mechanism of mitochondrial free radical generation due to a compromised mitochondrial antioxidant system caused by intra-mitochondrial NAD+ depletion. Finally, the interplay between different mechanisms of mitochondrial ROS generation and potential therapeutic approaches are reviewed.
Keywords: NAD+; acetylation; free radicals; ischemia; mitochondria.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
