

Novel ECHS1 mutations in Leigh syndrome identified by whole-exome sequencing in five Chinese families: case report
- PMID: 32677908
- PMCID: PMC7366304
- DOI: 10.1186/s12881-020-01083-1
Novel ECHS1 mutations in Leigh syndrome identified by whole-exome sequencing in five Chinese families: case report
Abstract
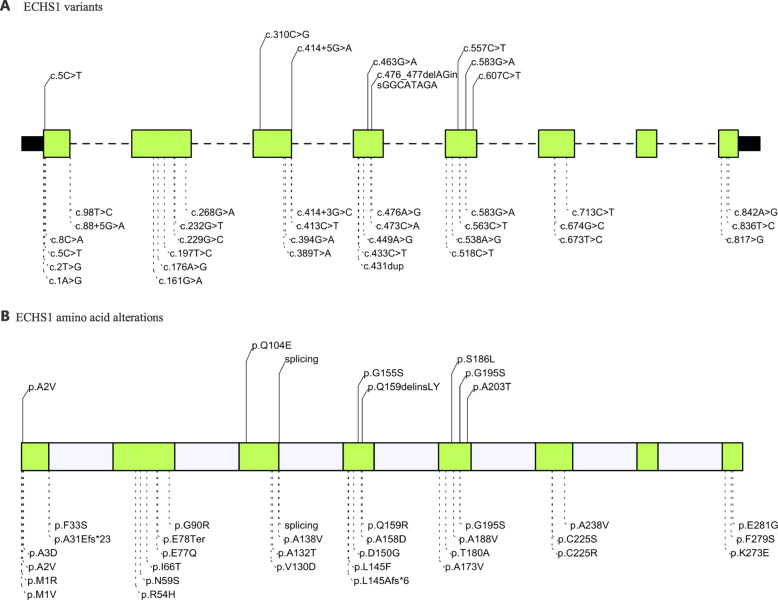
Background: Short-chain enoyl-CoA hydratase deficiency (ECHS1D), also known as ECHS1 deficiency, is a rare inborn metabolic disorder with clinical presentations characterized by Leigh syndrome (LS). Thirty-four different pathogenic mutations have been identified from over 40 patients to date.
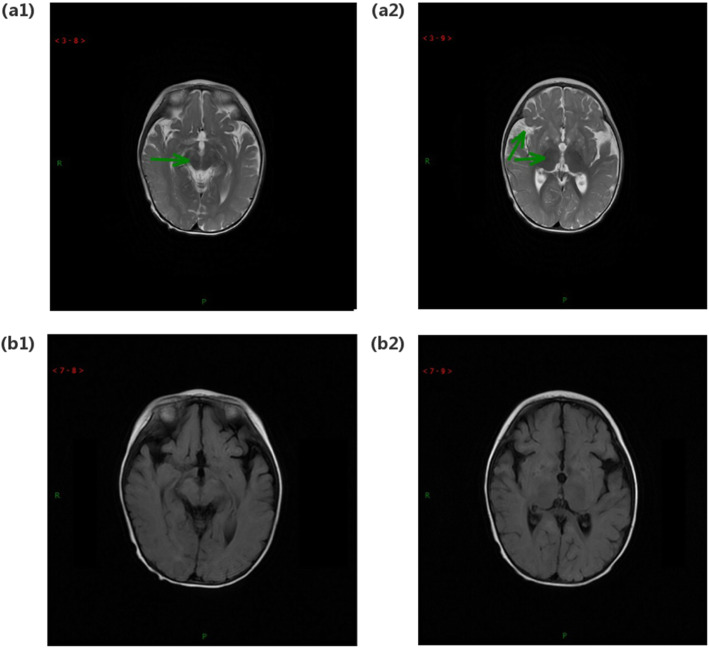
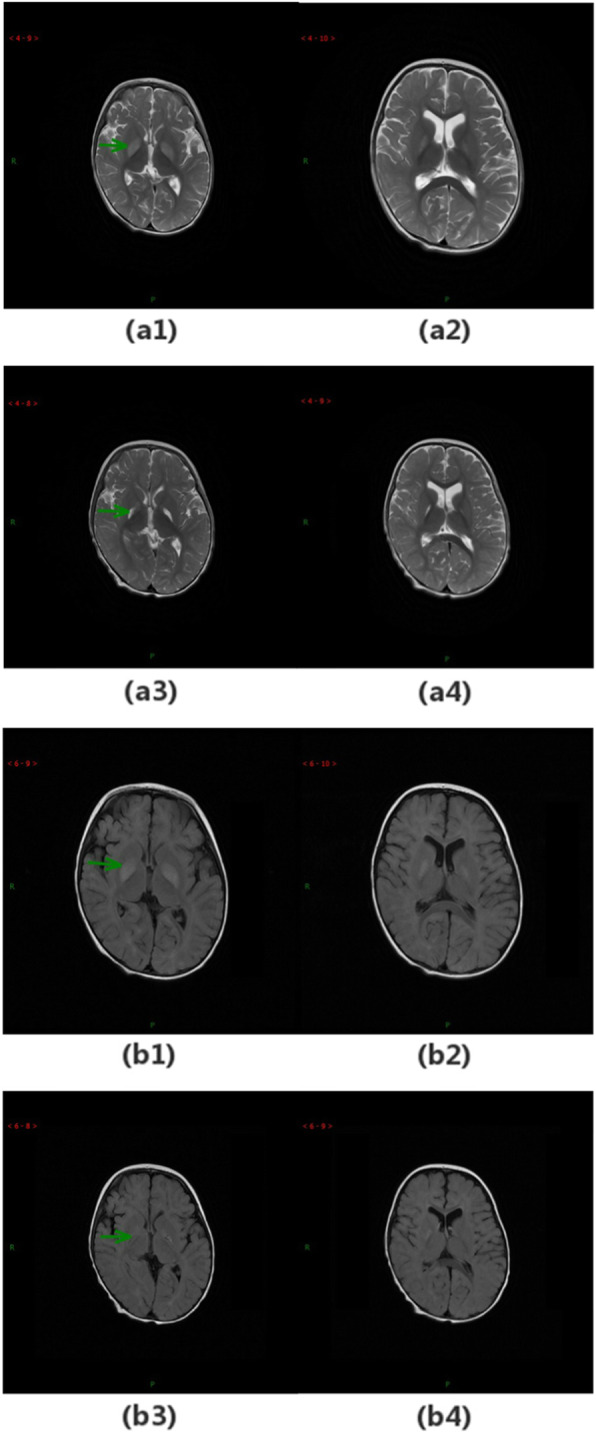
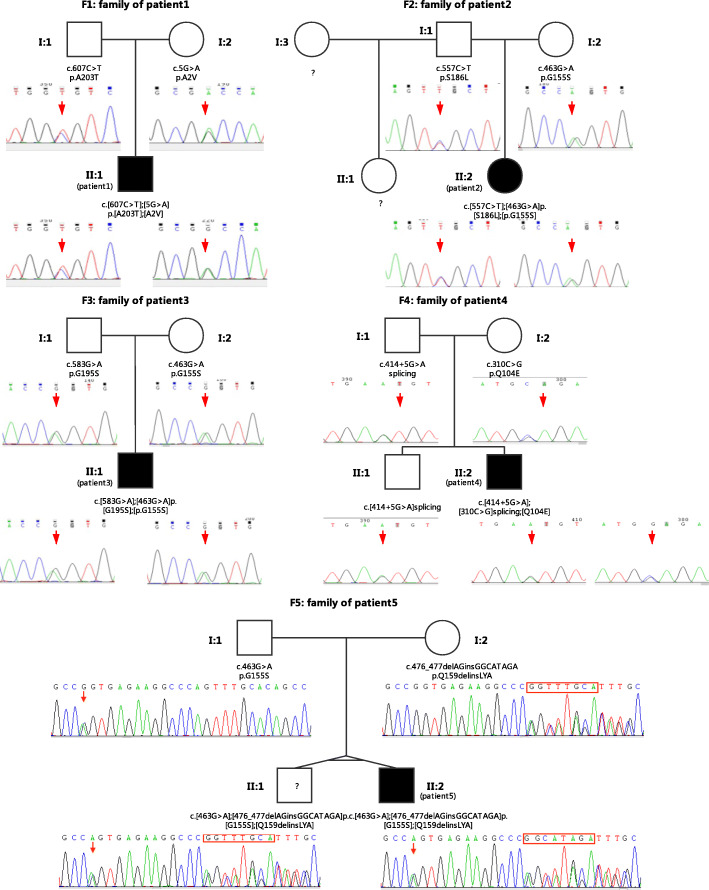
Case presentation: Here, we report five Chinese patients with clinical syndromes typified as LS. Despite different initial symptoms, all patients presented developmental regression, dystonia, common radiological features such as symmetrical bilateral brain abnormalities, and similar metabolic results such as elevated plasma lactate and 2,3-dihydroxy-2-methylbutyrate. Utilizing whole-exome sequencing (WES), we identified eight distinct variants in ECHS1, with six novel variants, and the remaining two variants have been previously reported. Interestingly, one of the six novel variants, c.463G > A (p.Gly155Ser), was detected in three patients from unrelated families, suggesting a potential founder effect already described for a few mutations in LS. Incorporating both genetic analysis and medical results, including magnetic resonance imaging (MRI), electroencephalography (EEG), and biochemical testing, our study enriched the mutation spectrum of the ECHS1 gene and confirmed the phenotypic presentations of LS.
Conclusions: The severity of ECHS1 deficiency seems to vary. It was affected by both genetics and external environmental factors that lead to increased metabolism. Our study enriched the mutation spectrum of the ECHS1 gene, confirmed the phenotypic presentations, and highlighted the importance of the valine catabolic pathway in Leigh syndrome. Further studies are required to examine the potential founder mutation c.463G > A (p.Gly155Ser) and the role of ECHS1 in relevant pathways.
Keywords: Case report; ECHS1; Leigh syndrome; Whole-exome sequencing.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Clinical, biochemical, and genetic features of four patients with short-chain enoyl-CoA hydratase (ECHS1) deficiency.Am J Med Genet A. 2018 May;176(5):1115-1127. doi: 10.1002/ajmg.a.38658. Epub 2018 Mar 25. Am J Med Genet A. 2018. PMID: 29575569 Free PMC article.
-
Whole-exome sequencing identifies novel ECHS1 mutations in Leigh syndrome.Hum Genet. 2015 Sep;134(9):981-91. doi: 10.1007/s00439-015-1577-y. Epub 2015 Jun 23. Hum Genet. 2015. PMID: 26099313
-
Clinical, biochemical and metabolic characterization of patients with short-chain enoyl-CoA hydratase(ECHS1) deficiency: two case reports and the review of the literature.BMC Pediatr. 2020 Feb 3;20(1):50. doi: 10.1186/s12887-020-1947-z. BMC Pediatr. 2020. PMID: 32013919 Free PMC article. Review.
-
ECHS1 mutations in Leigh disease: a new inborn error of metabolism affecting valine metabolism.Brain. 2014 Nov;137(Pt 11):2903-8. doi: 10.1093/brain/awu216. Epub 2014 Aug 14. Brain. 2014. PMID: 25125611
-
Pathogenic Biallelic Mutations in ECHS1 in a Case with Short-Chain Enoyl-CoA Hydratase (SCEH) Deficiency-Case Report and Literature Review.Int J Environ Res Public Health. 2022 Feb 13;19(4):2088. doi: 10.3390/ijerph19042088. Int J Environ Res Public Health. 2022. PMID: 35206276 Free PMC article. Review.
Cited by
-
Time-resolved mitochondrial screen identifies regulatory components of oxidative metabolism.EMBO Rep. 2025 Jun;26(12):3045-3074. doi: 10.1038/s44319-025-00459-9. Epub 2025 Apr 29. EMBO Rep. 2025. PMID: 40301572 Free PMC article.
-
Exploring triheptanoin as treatment for short chain enoyl CoA hydratase deficiency.Ann Clin Transl Neurol. 2021 May;8(5):1151-1157. doi: 10.1002/acn3.51359. Epub 2021 May 1. Ann Clin Transl Neurol. 2021. PMID: 33931985 Free PMC article.
-
Histone crotonylation in neurobiology: to be or not to be?Chin Med J (Engl). 2022 May 5;135(9):1036-1038. doi: 10.1097/CM9.0000000000001945. Chin Med J (Engl). 2022. PMID: 35067563 Free PMC article. No abstract available.
-
Structural and biochemical mechanism of short-chain enoyl-CoA hydratase (ECHS1) substrate recognition.Commun Biol. 2025 Apr 16;8(1):619. doi: 10.1038/s42003-025-07924-0. Commun Biol. 2025. PMID: 40240482 Free PMC article.
-
ECHS1: pathogenic mechanisms, experimental models, and emerging therapeutic strategies.Orphanet J Rare Dis. 2025 Aug 13;20(1):430. doi: 10.1186/s13023-025-03959-y. Orphanet J Rare Dis. 2025. PMID: 40804397 Free PMC article. Review.
References
-
- Ferdinandusse S, Waterham HR, Heales SJ, Brown GK, Hargreaves IP, Taanman J-W, et al. HIBCH mutations can cause Leigh-like disease with combined deficiency of multiple mitochondrial respiratory chain enzymes and pyruvate dehydrogenase. Orphanet J Rare Dis. 2013;8(1):188. doi: 10.1186/1750-1172-8-188. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
