

Genotype-Phenotype Correlation in Children: The Impact of FBN1 Variants on Pediatric Marfan Care
- PMID: 32679894
- PMCID: PMC7397236
- DOI: 10.3390/genes11070799
Genotype-Phenotype Correlation in Children: The Impact of FBN1 Variants on Pediatric Marfan Care
Abstract
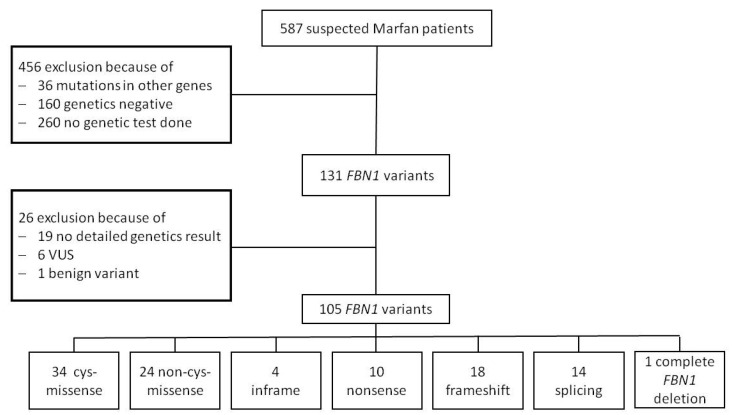
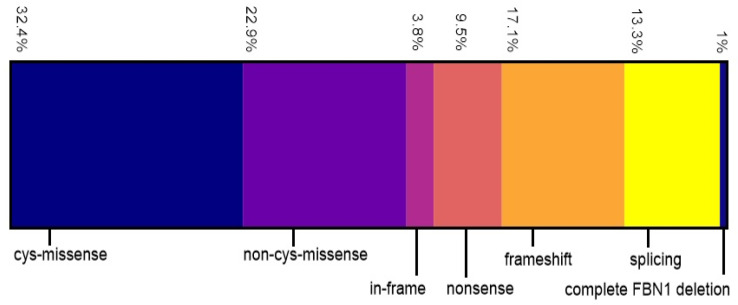
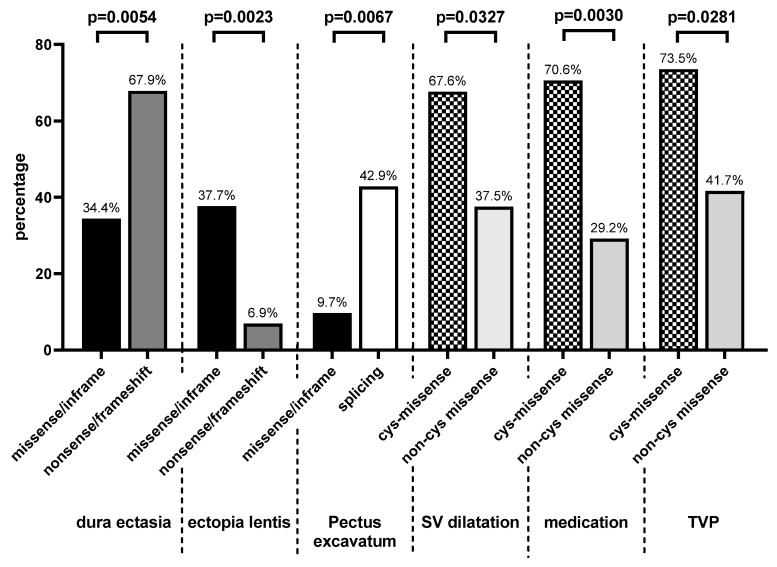
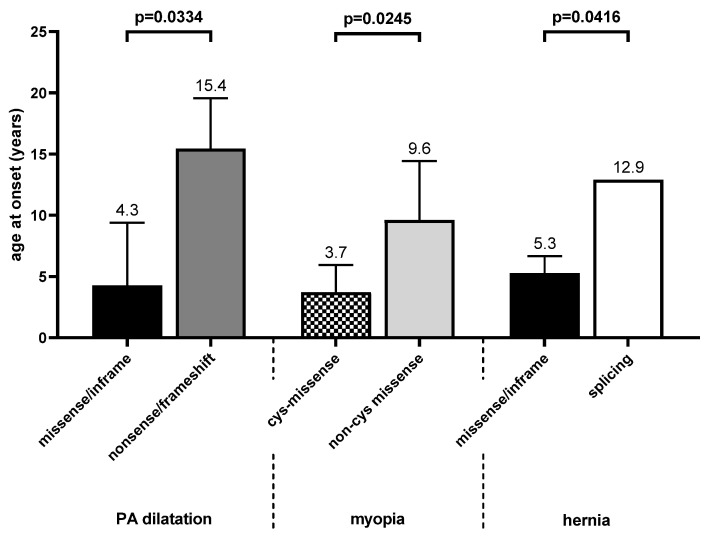
Currently, no reliable genotype-phenotype correlation is available for pediatric Marfan patients in everyday clinical practice. We investigated correlations of FBN1 variants with the prevalence and age of onset of Marfan manifestations in childhood and differentiated three groups: missense/in-frame, splice, and nonsense/frameshift variants. In addition, we differentiated missense variants destroying or generating a cysteine (cys-missense) and alterations not affecting cysteine. We categorized 105 FBN1-positive pediatric patients. Patients with cys-missense more frequently developed aortic dilatation (p = 0.03) requiring medication (p = 0.003), tricuspid valve prolapse (p = 0.03), and earlier onset of myopia (p = 0.02) than those with other missense variants. Missense variants correlated with a higher prevalence of ectopia lentis (p = 0.002) and earlier onset of pulmonary artery dilatation (p = 0.03) than nonsense/frameshift, and dural ectasia was more common in the latter (p = 0.005). Pectus excavatum (p = 0.007) appeared more often in patients with splice compared with missense/in-frame variants, while hernia (p = 0.04) appeared earlier in the latter. Findings on genotype-phenotype correlations in Marfan-affected children can improve interdisciplinary therapy. In patients with cys-missense variants, early medical treatment of aortic dilatation seems reasonable and early regular ophthalmologic follow-up essential. Patients with nonsense/frameshift and splice variants require early involvement of orthopedic specialists to support the growing child.
Keywords: FBN1 variant; Marfan syndrome; childhood; genetic testing; genotype–phenotype; variant spectrum.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Arnaud P., Hanna N., Aubart M., Leheup B., Dupuis-Girod S., Naudion S., Lacombe D., Milleron O., Odent S., Faivre L., et al. Homozygous and compound heterozygous mutations in the FBN1 gene: Unexpected findings in molecular diagnosis of Marfan syndrome. J. Med. Genet. 2017;54:100–103. doi: 10.1136/jmedgenet-2016-103996. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
