

Cross-species regulatory sequence activity prediction
- PMID: 32687525
- PMCID: PMC7392335
- DOI: 10.1371/journal.pcbi.1008050
Cross-species regulatory sequence activity prediction
Abstract
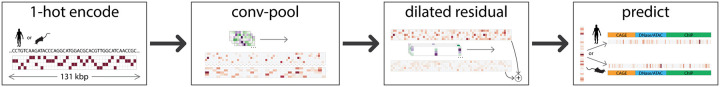
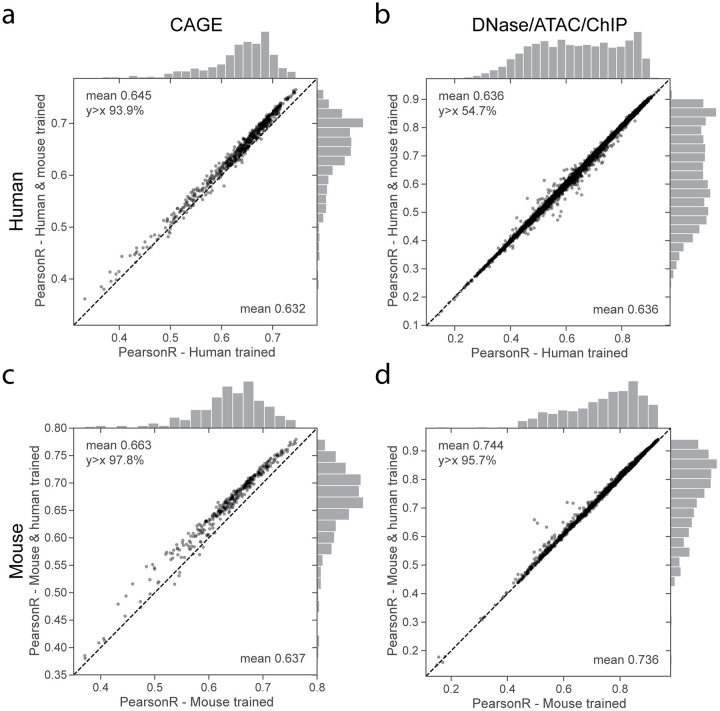
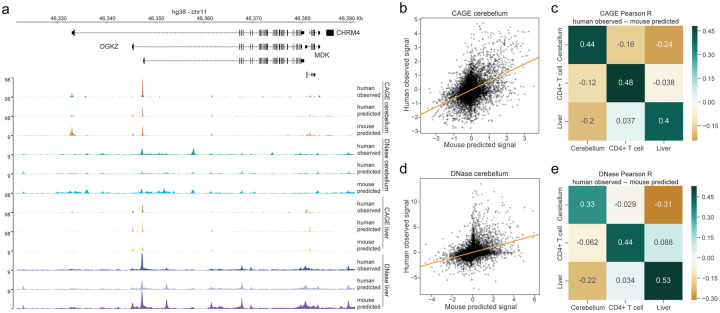
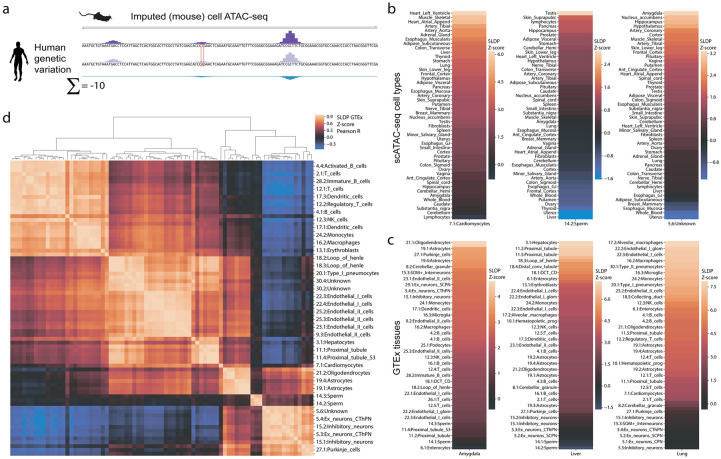
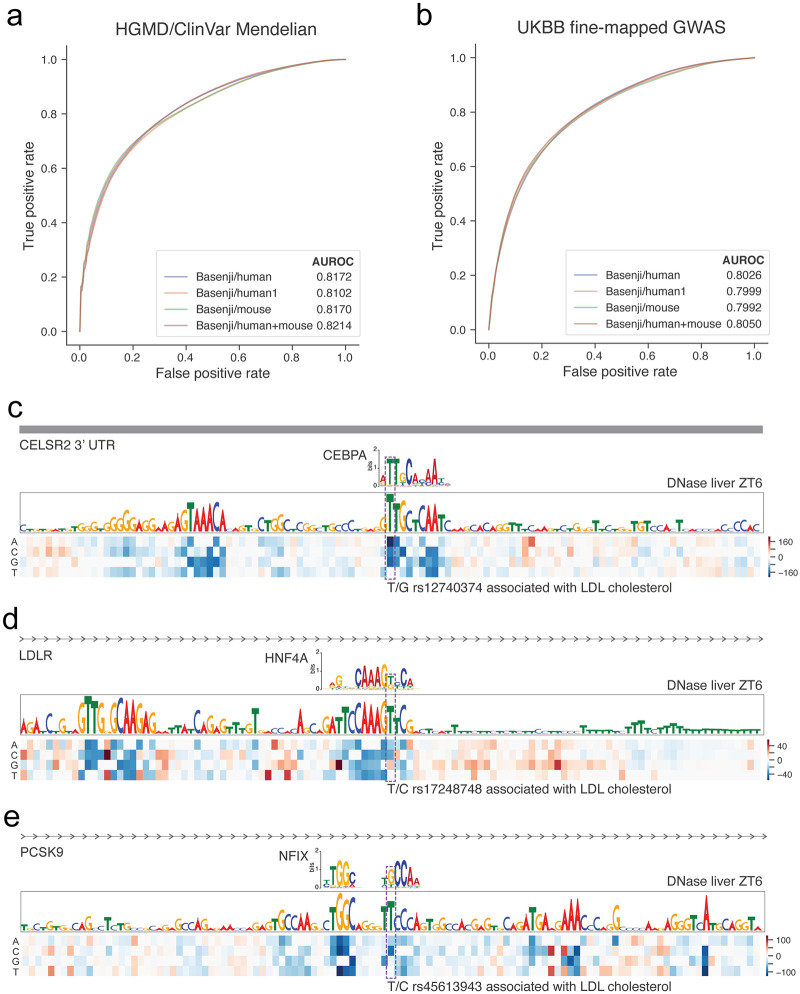
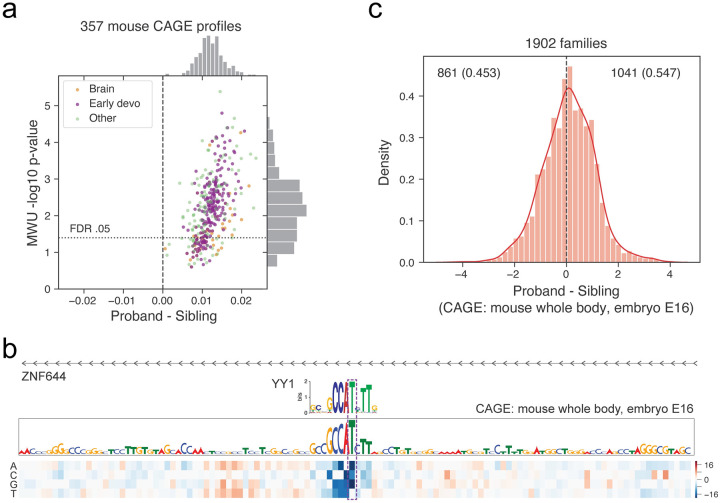
Machine learning algorithms trained to predict the regulatory activity of nucleic acid sequences have revealed principles of gene regulation and guided genetic variation analysis. While the human genome has been extensively annotated and studied, model organisms have been less explored. Model organism genomes offer both additional training sequences and unique annotations describing tissue and cell states unavailable in humans. Here, we develop a strategy to train deep convolutional neural networks simultaneously on multiple genomes and apply it to learn sequence predictors for large compendia of human and mouse data. Training on both genomes improves gene expression prediction accuracy on held out and variant sequences. We further demonstrate a novel and powerful approach to apply mouse regulatory models to analyze human genetic variants associated with molecular phenotypes and disease. Together these techniques unleash thousands of non-human epigenetic and transcriptional profiles toward more effective investigation of how gene regulation affects human disease.
Conflict of interest statement
DRK is employed by Calico LLC.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
