

Insights into Autosomal Dominant Polycystic Kidney Disease from Genetic Studies
- PMID: 32690722
- PMCID: PMC8259493
- DOI: 10.2215/CJN.02320220
Insights into Autosomal Dominant Polycystic Kidney Disease from Genetic Studies
Abstract
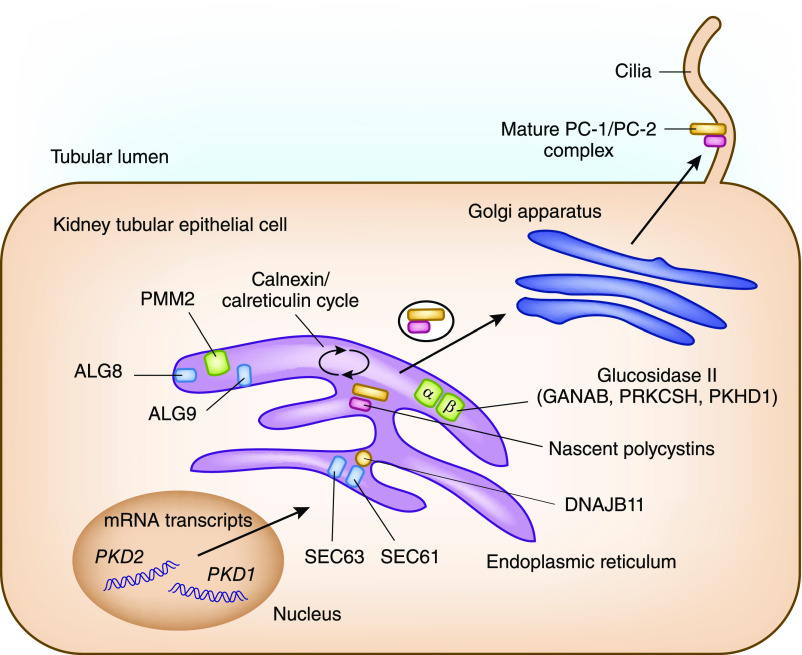
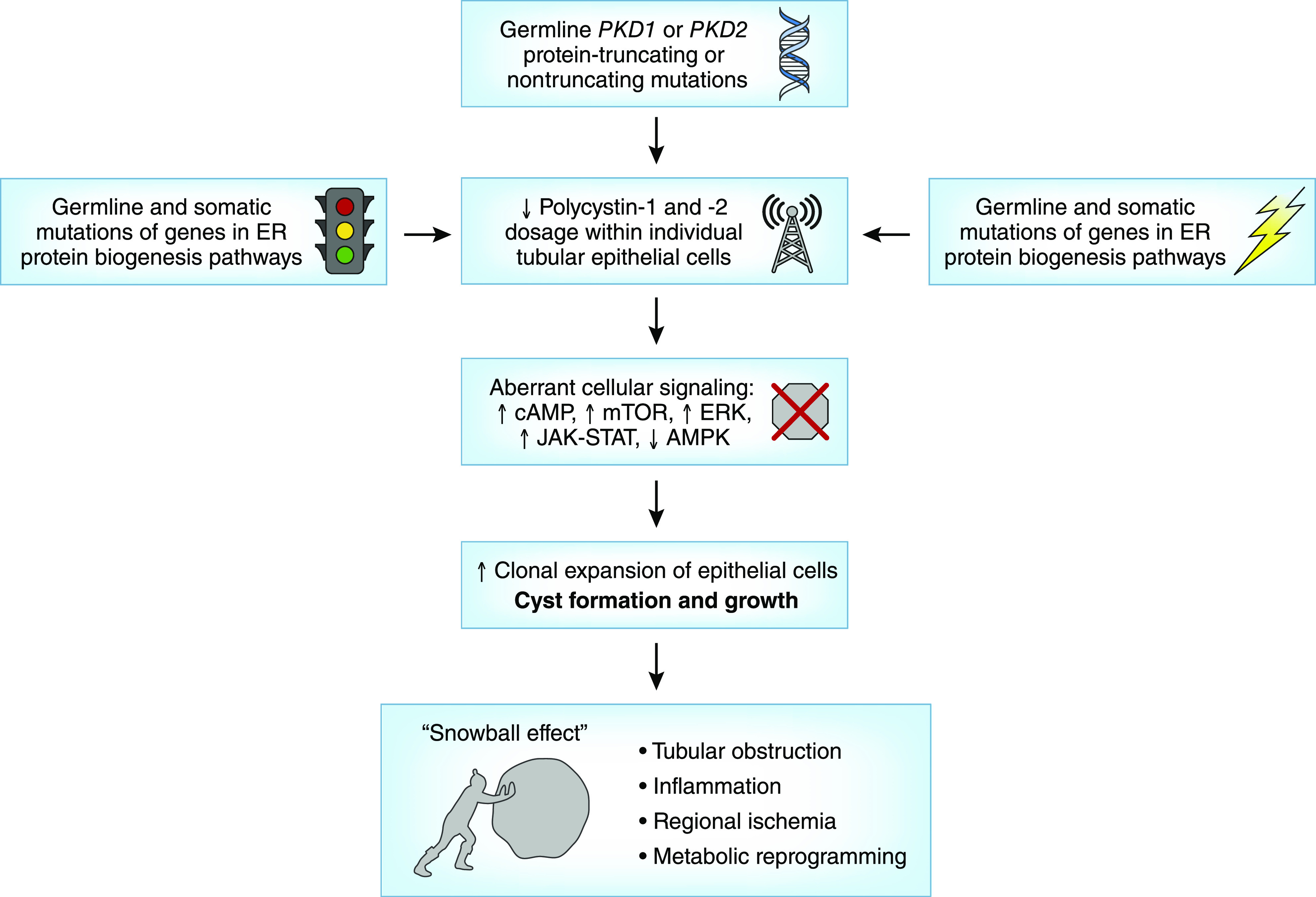
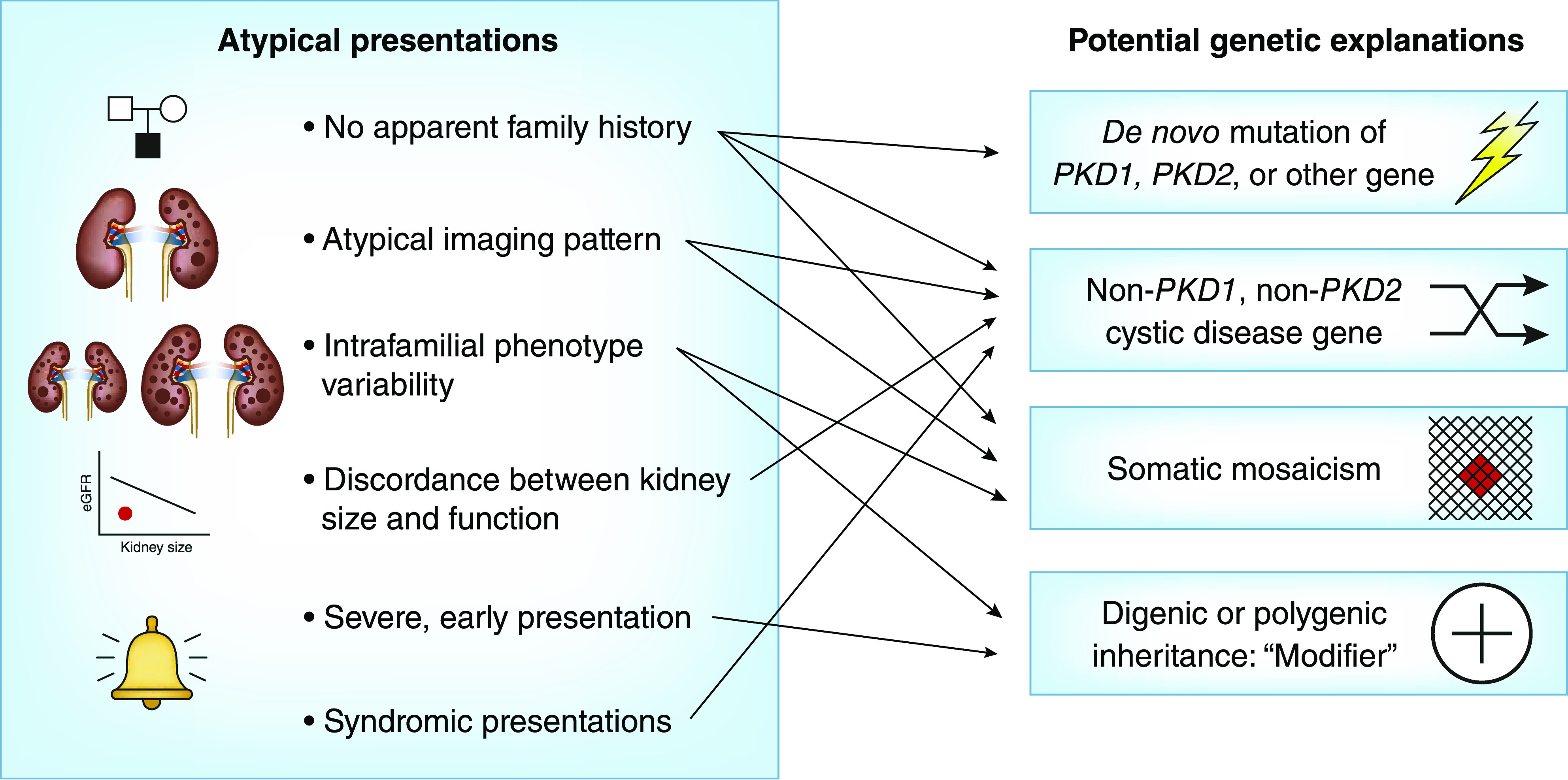
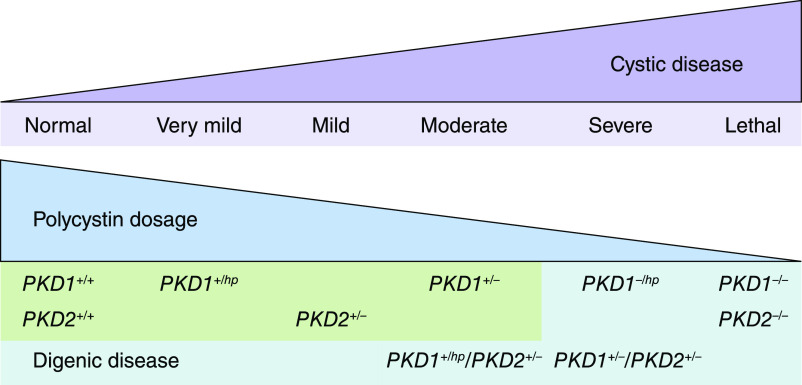
Autosomal dominant polycystic kidney disease is the most common monogenic cause of ESKD. Genetic studies from patients and animal models have informed disease pathobiology and strongly support a "threshold model" in which cyst formation is triggered by reduced functional polycystin dosage below a critical threshold within individual tubular epithelial cells due to (1) germline and somatic PKD1 and/or PKD2 mutations, (2) mutations of genes (e.g., SEC63, SEC61B, GANAB, PRKCSH, DNAJB11, ALG8, and ALG9) in the endoplasmic reticulum protein biosynthetic pathway, or (3) somatic mosaicism. Genetic testing has the potential to provide diagnostic and prognostic information in cystic kidney disease. However, mutation screening of PKD1 is challenging due to its large size and complexity, making it both costly and labor intensive. Moreover, conventional Sanger sequencing-based genetic testing is currently limited in elucidating the causes of atypical polycystic kidney disease, such as within-family disease discordance, atypical kidney imaging patterns, and discordant disease severity between total kidney volume and rate of eGFR decline. In addition, environmental factors, genetic modifiers, and somatic mosaicism also contribute to disease variability, further limiting prognostication by mutation class in individual patients. Recent innovations in next-generation sequencing are poised to transform and extend molecular diagnostics at reasonable costs. By comprehensive screening of multiple cystic disease and modifier genes, targeted gene panel, whole-exome, or whole-genome sequencing is expected to improve both diagnostic and prognostic accuracy to advance personalized medicine in autosomal dominant polycystic kidney disease.
Keywords: Kidney Genomics Series; polycystic kidney disease.
Copyright © 2021 by the American Society of Nephrology.
Figures
Comment in
-
A Patient Perspective on Genetic Testing for ADPKD: The Lack of Complete Genetic Information, Especially Early in the Course of the Disease, Is Harming Adult Autosomal Dominant Polycystic Kidney Disease (ADPKD) Patients.Clin J Am Soc Nephrol. 2021 May 8;16(5):671-673. doi: 10.2215/CJN.14051119. Epub 2020 Jul 30. Clin J Am Soc Nephrol. 2021. PMID: 32732372 Free PMC article. No abstract available.
References
-
- Cornec-Le Gall E, Alam A, Perrone RD: Autosomal dominant polycystic kidney disease. Lancet 393: 919–935, 2019. - PubMed
-
- Lanktree MB, Chapman AB: New treatment paradigms for ADPKD: Moving towards precision medicine. Nat Rev Nephrol 13: 750–768, 2017. - PubMed
-
- Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millán JL, Gamble V, Harris PC: The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 10: 151–160, 1995. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
