

Prostate cancer reactivates developmental epigenomic programs during metastatic progression
- PMID: 32690948
- PMCID: PMC10007911
- DOI: 10.1038/s41588-020-0664-8
Prostate cancer reactivates developmental epigenomic programs during metastatic progression
Abstract
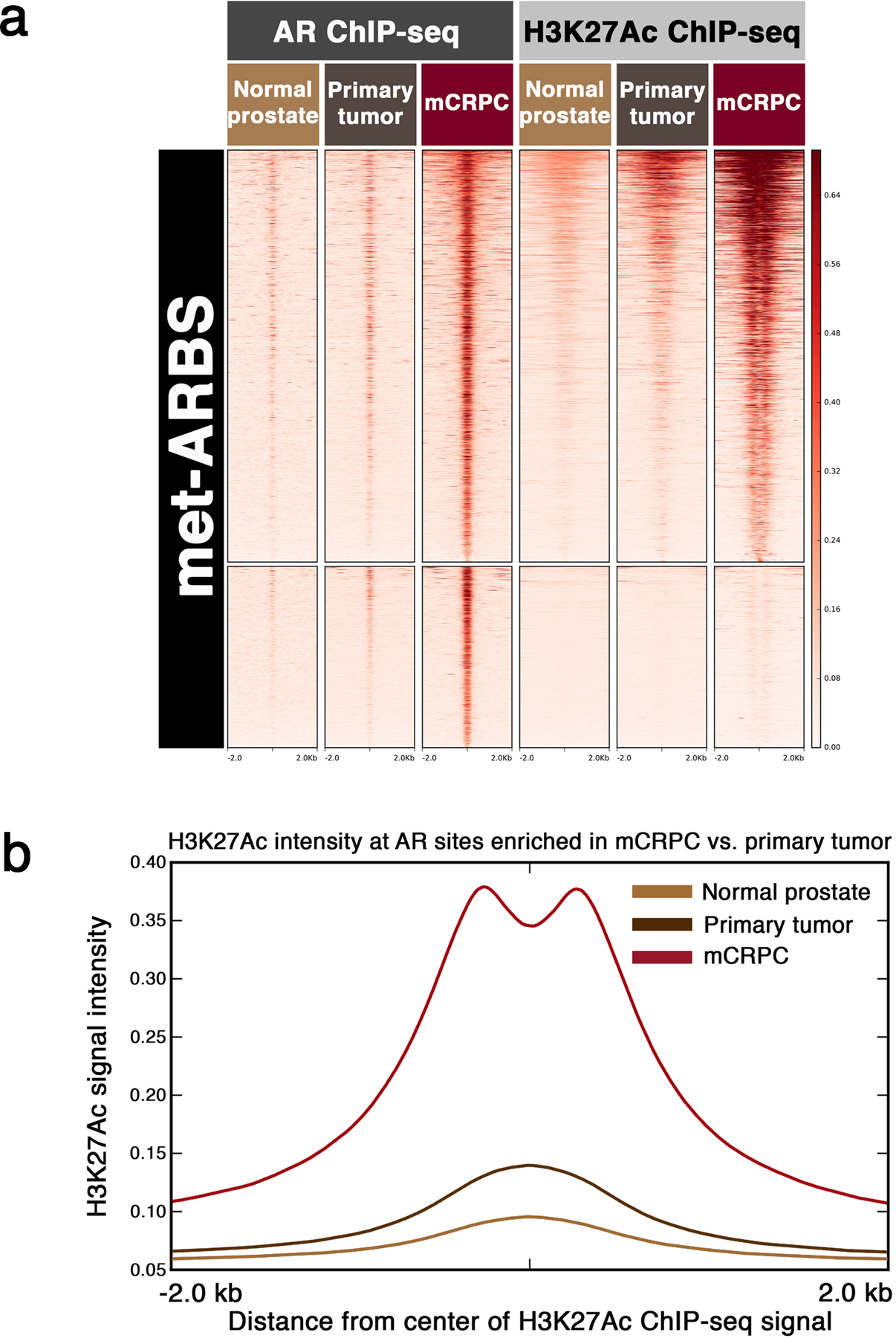
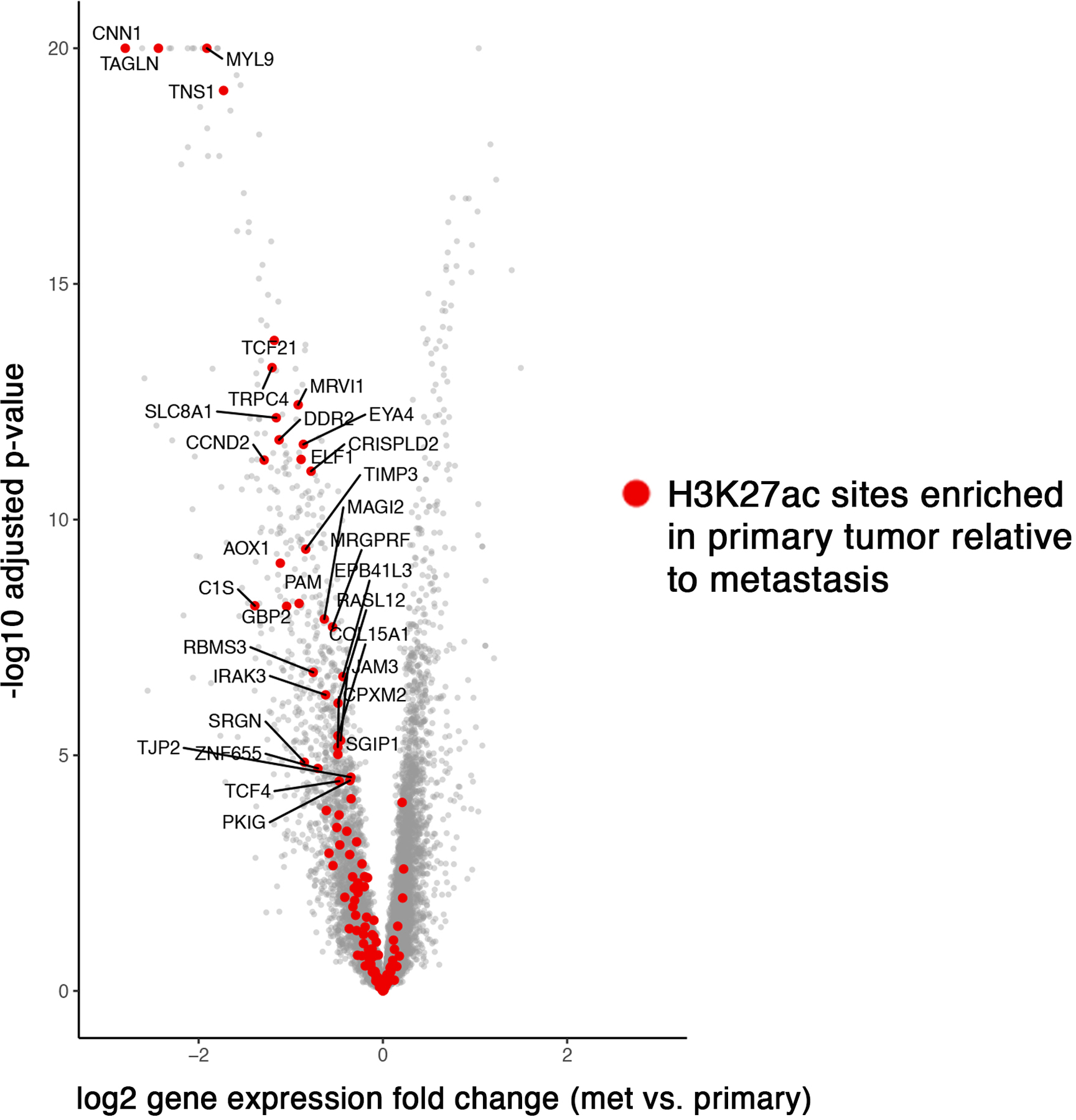
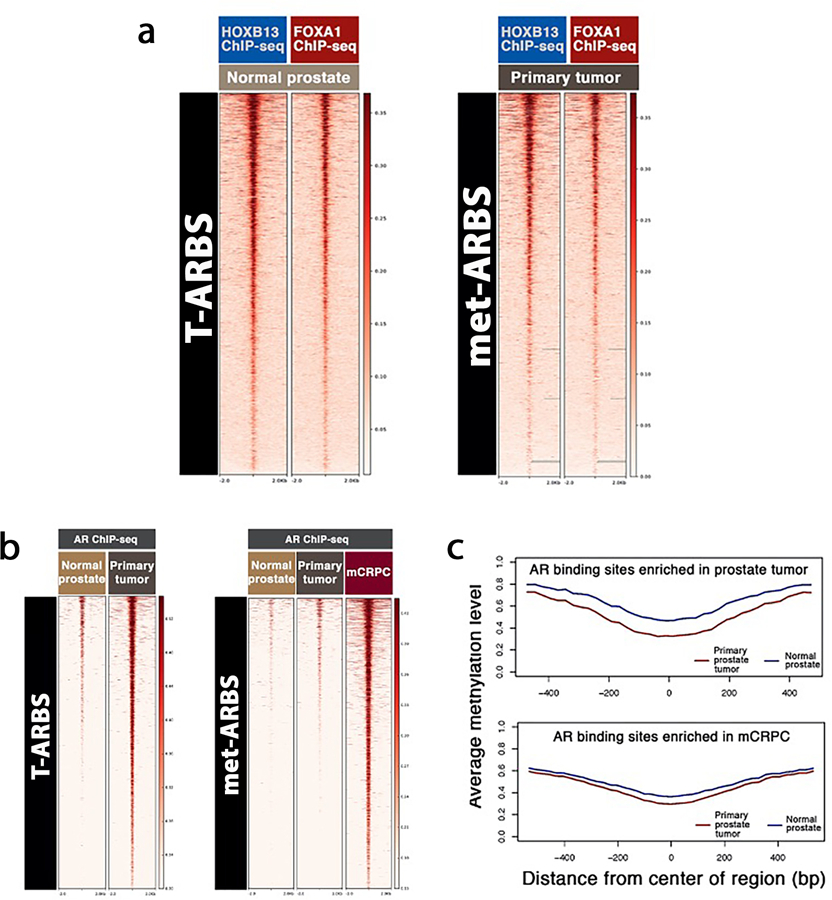
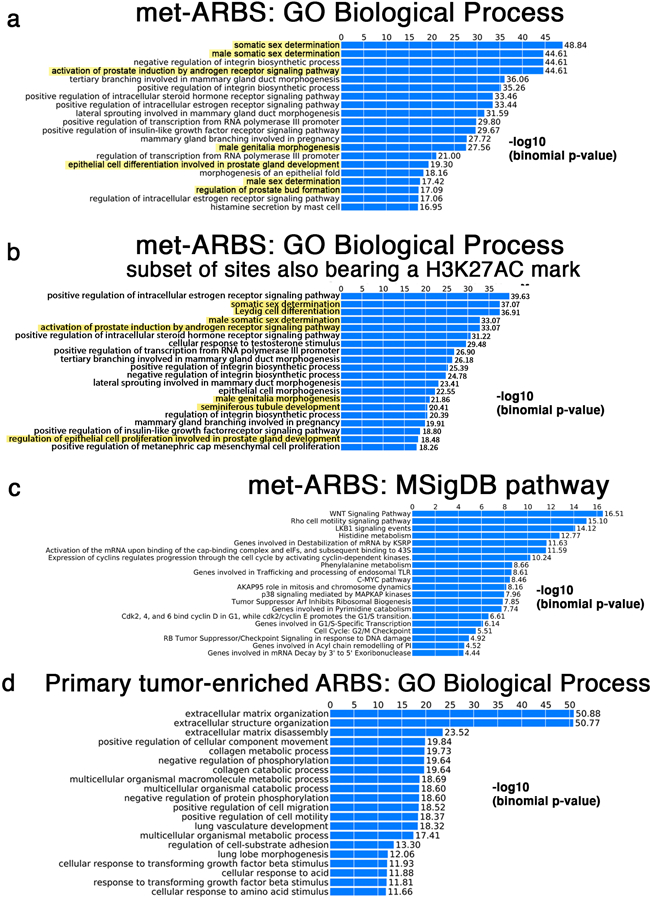
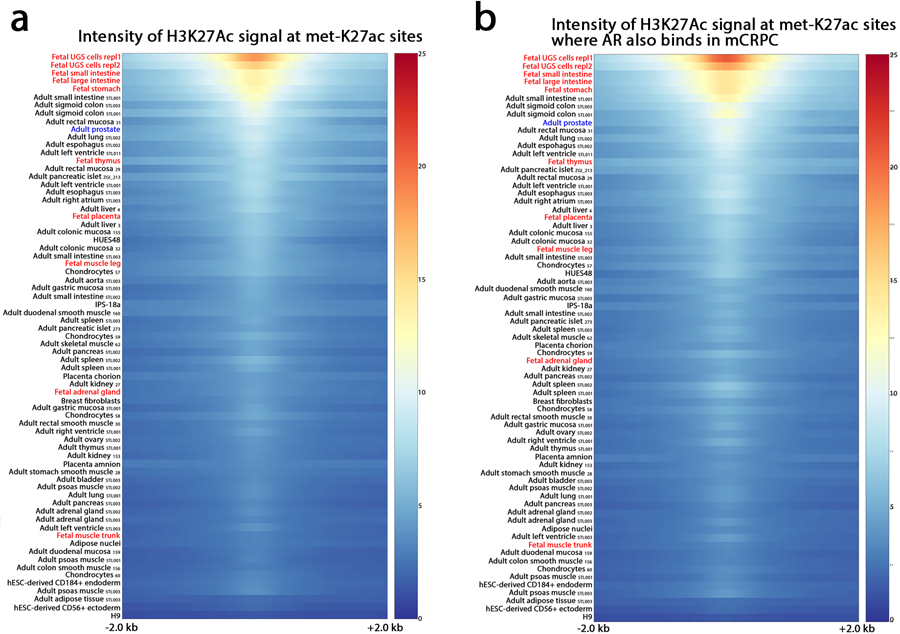
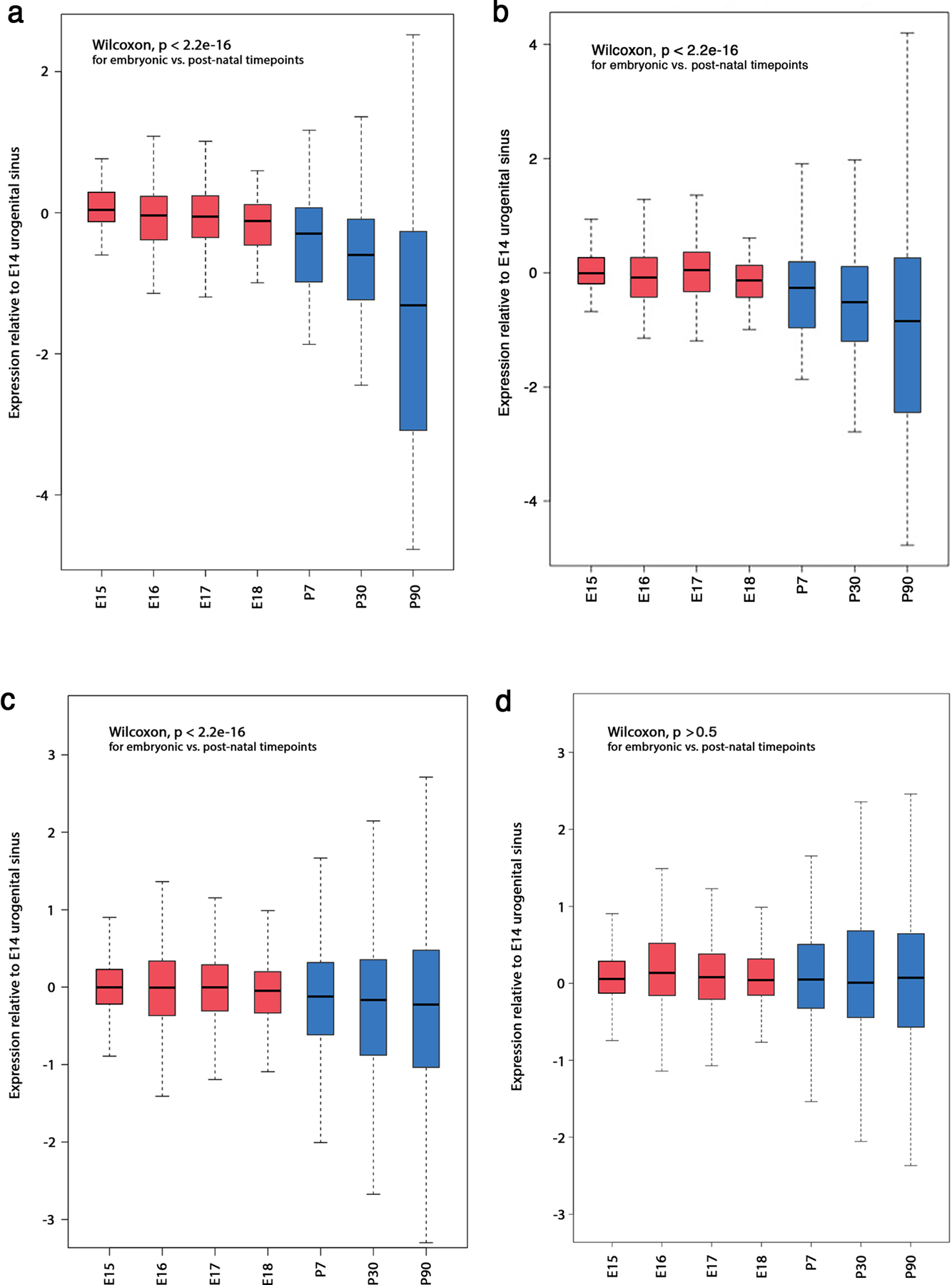
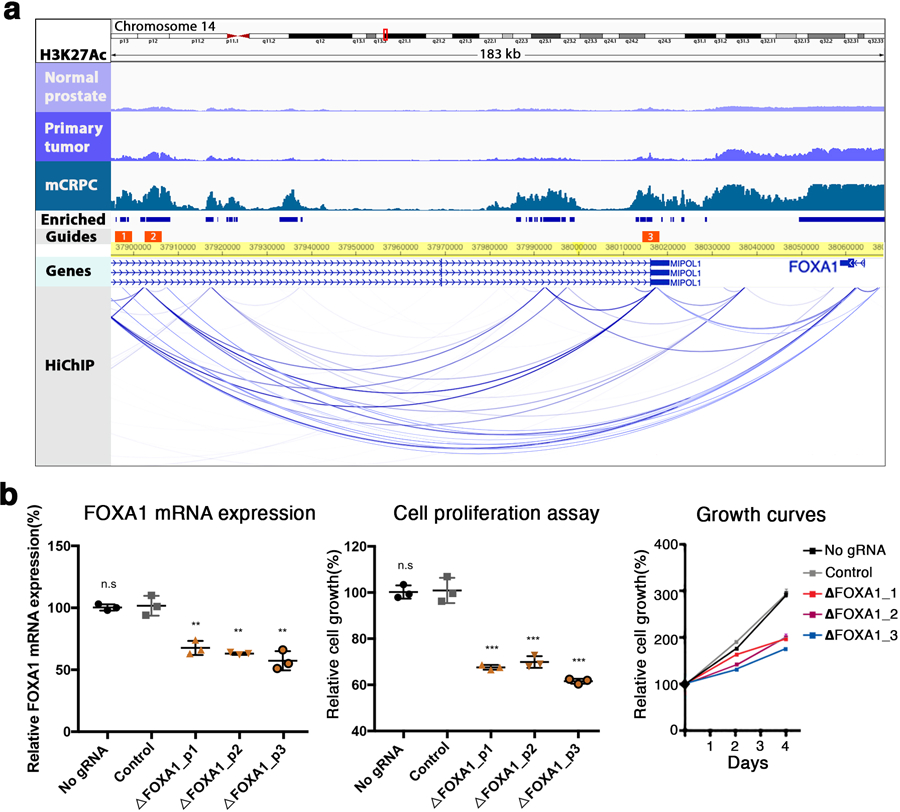
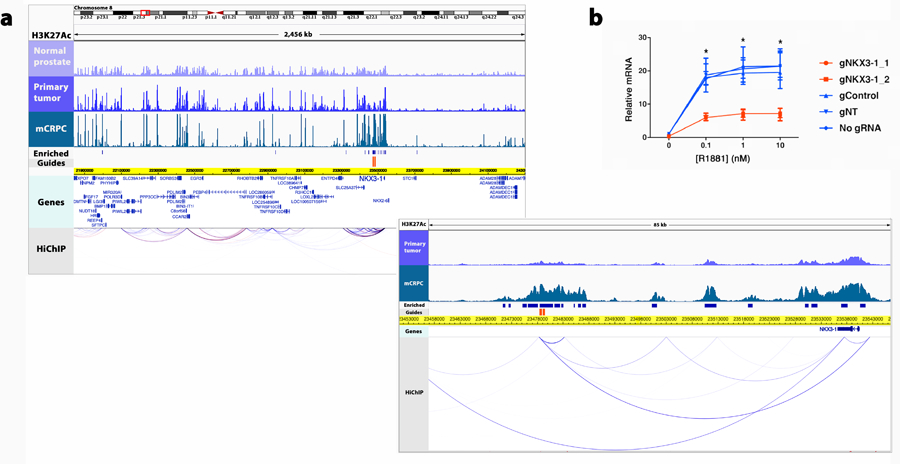
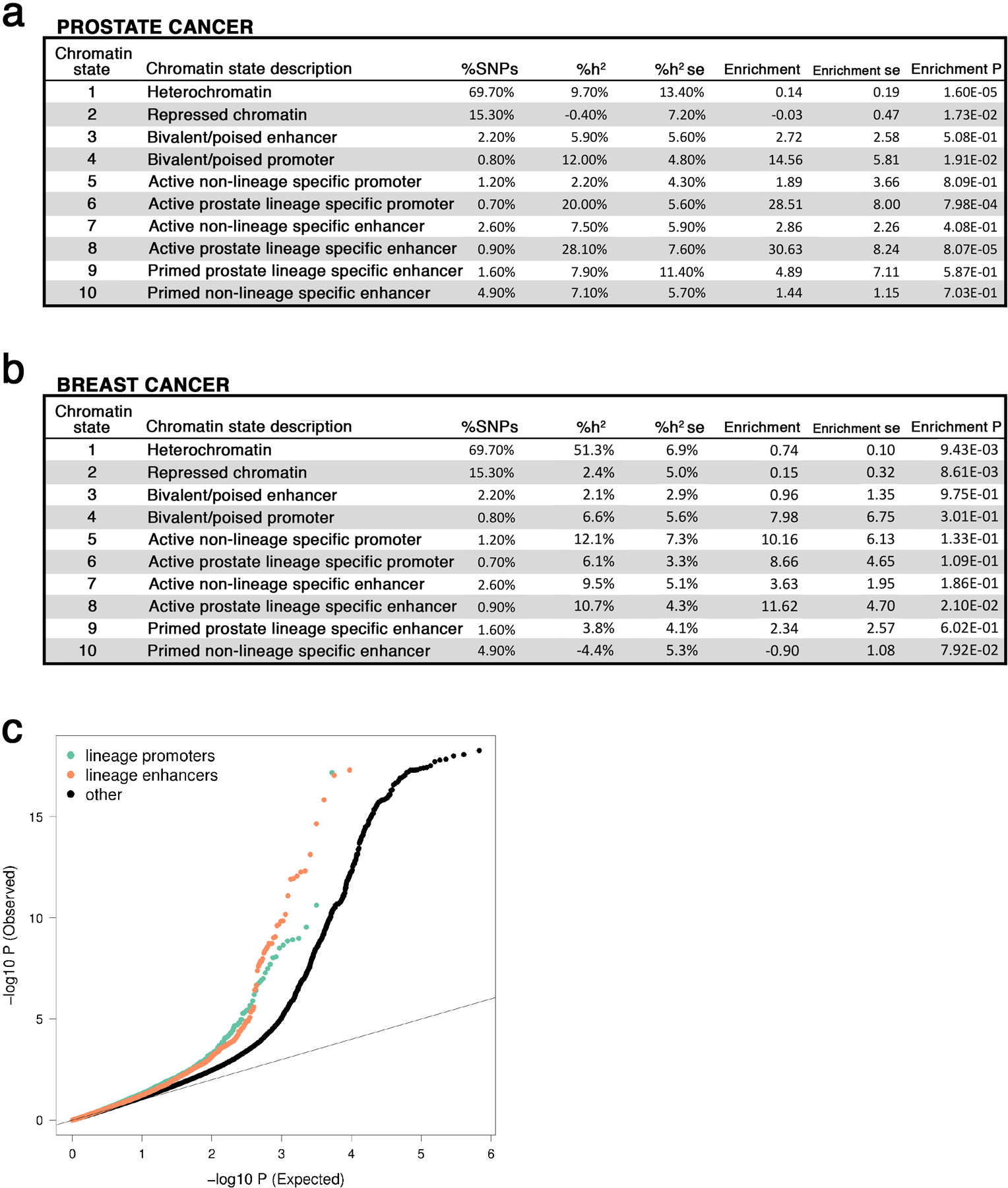
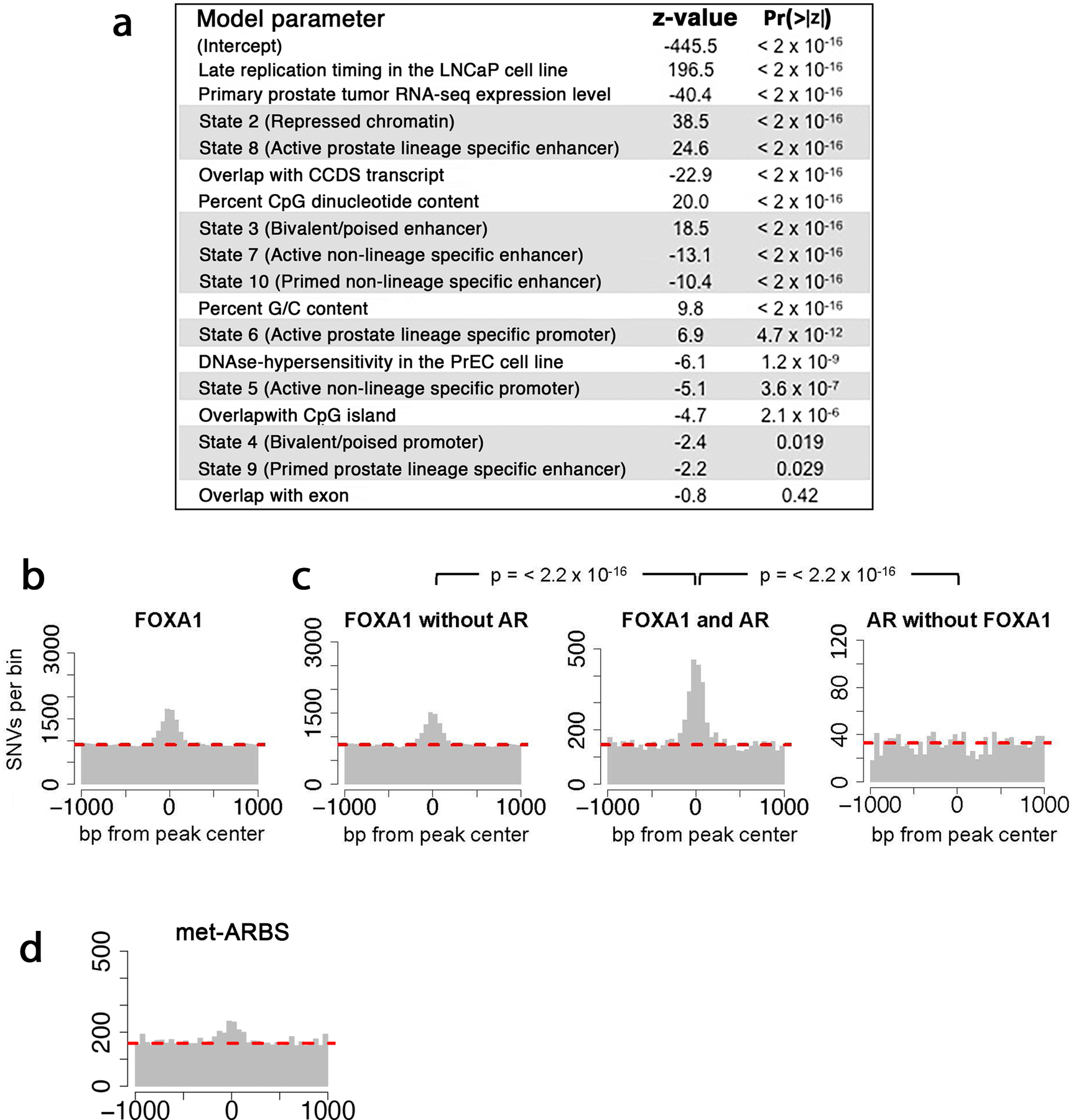
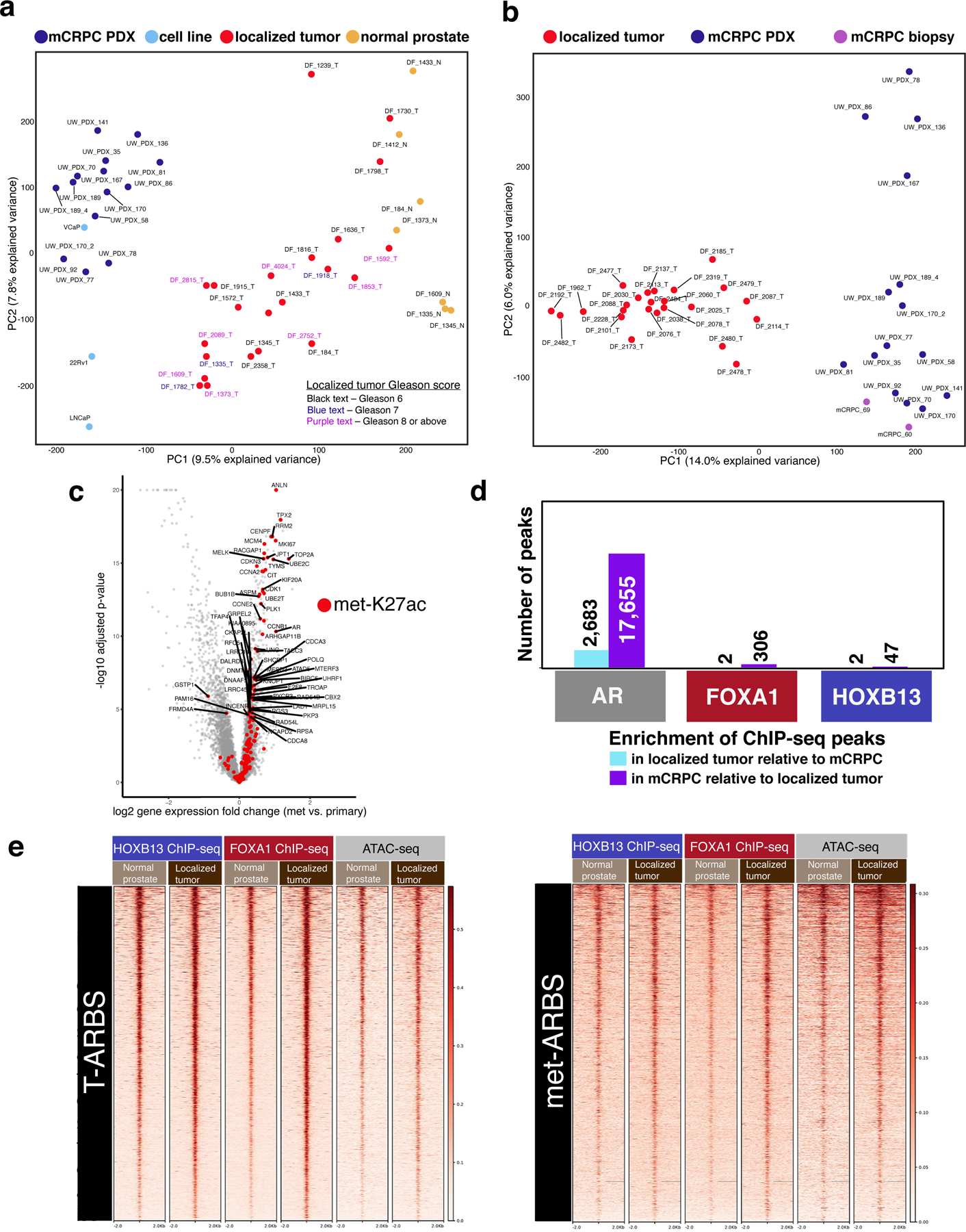
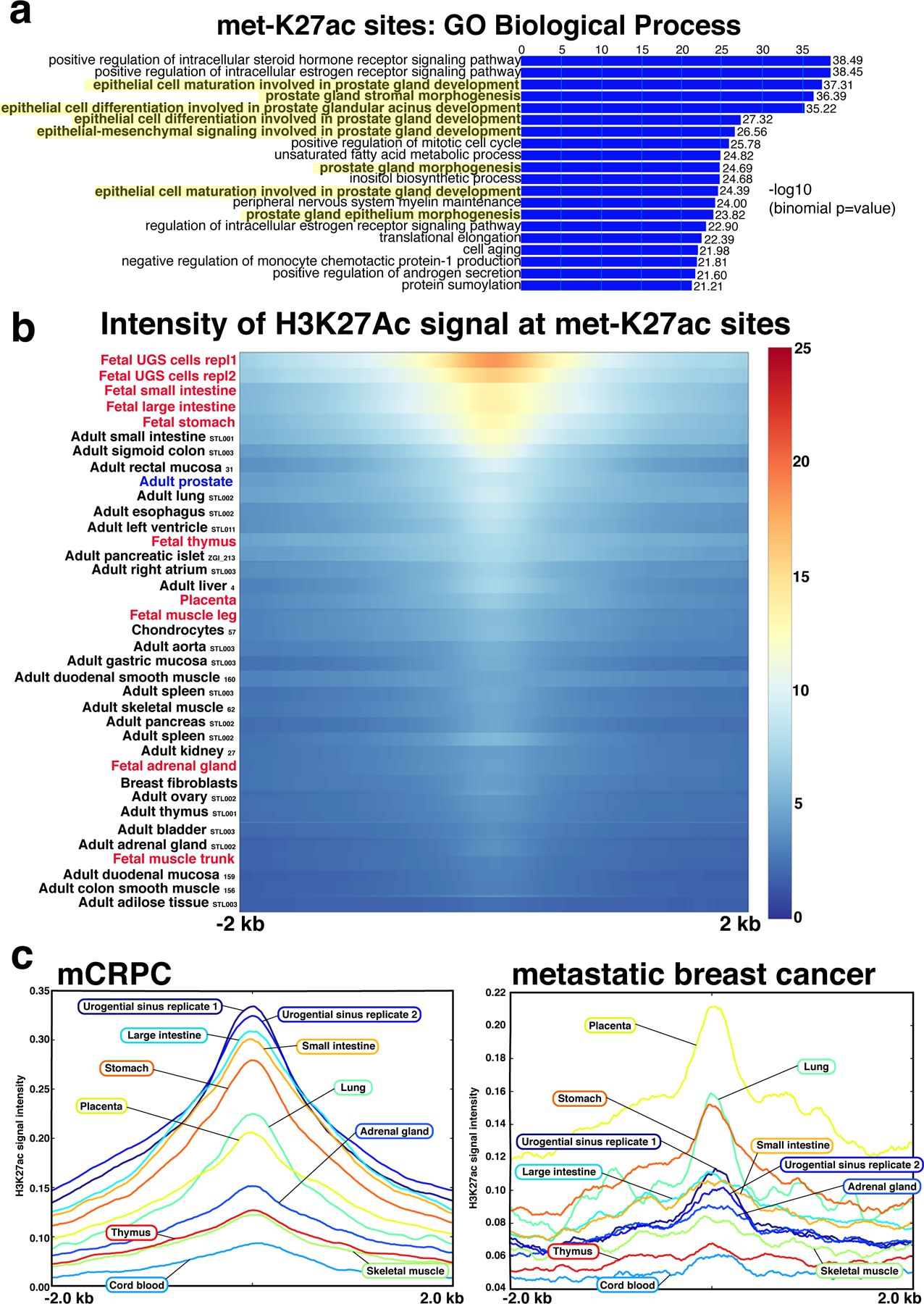
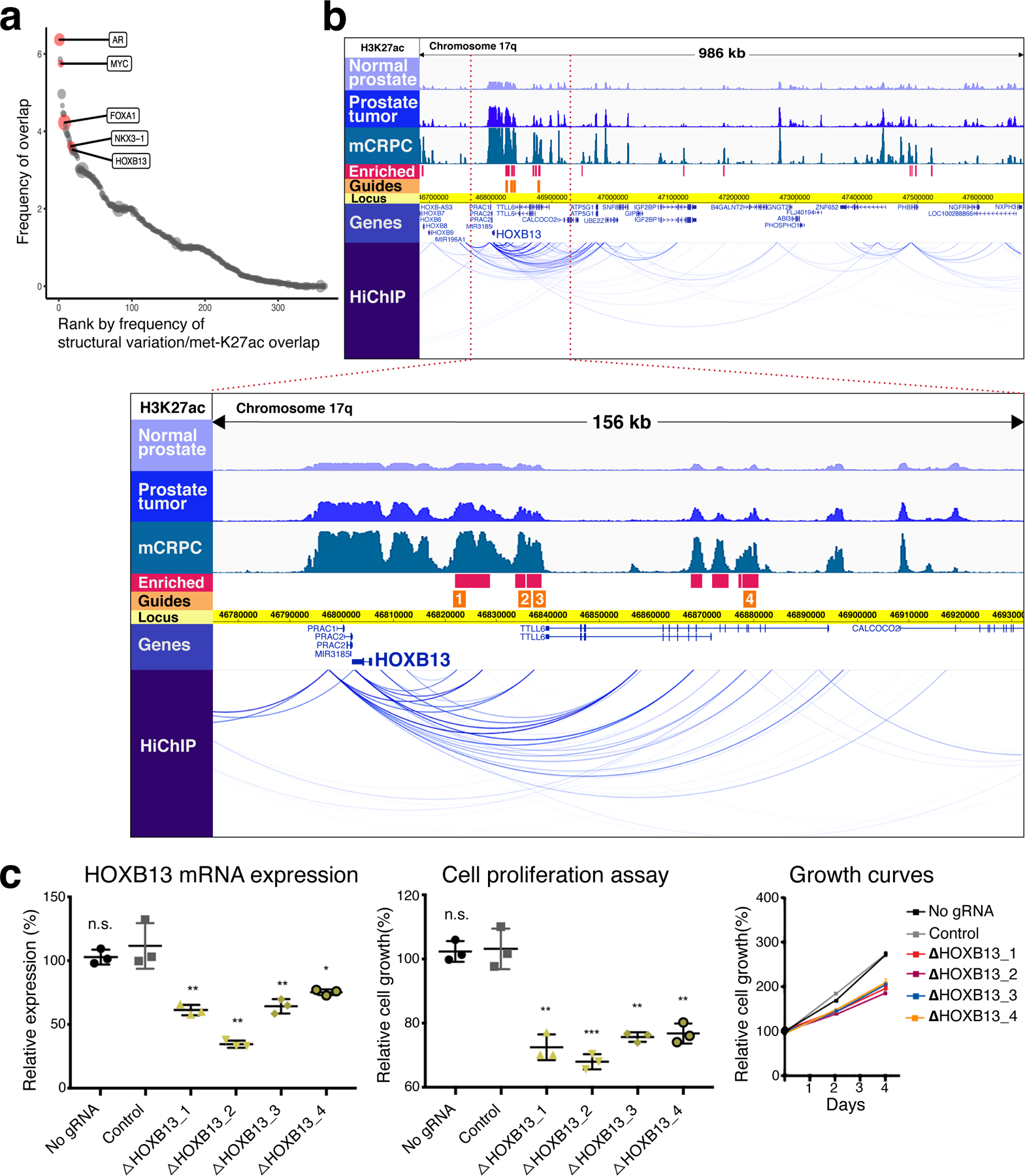
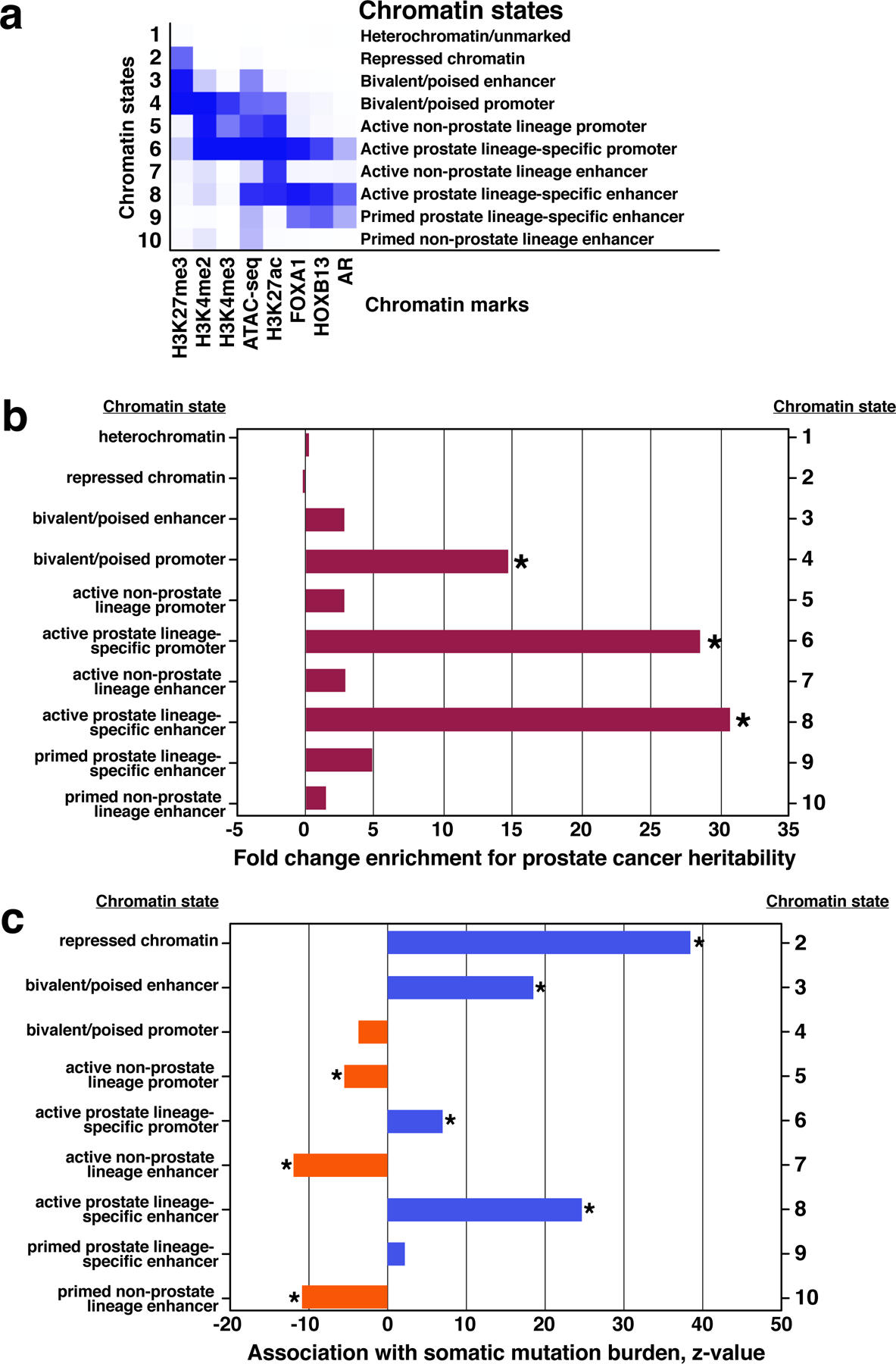
Epigenetic processes govern prostate cancer (PCa) biology, as evidenced by the dependency of PCa cells on the androgen receptor (AR), a prostate master transcription factor. We generated 268 epigenomic datasets spanning two state transitions-from normal prostate epithelium to localized PCa to metastases-in specimens derived from human tissue. We discovered that reprogrammed AR sites in metastatic PCa are not created de novo; rather, they are prepopulated by the transcription factors FOXA1 and HOXB13 in normal prostate epithelium. Reprogrammed regulatory elements commissioned in metastatic disease hijack latent developmental programs, accessing sites that are implicated in prostate organogenesis. Analysis of reactivated regulatory elements enabled the identification and functional validation of previously unknown metastasis-specific enhancers at HOXB13, FOXA1 and NKX3-1. Finally, we observed that prostate lineage-specific regulatory elements were strongly associated with PCa risk heritability and somatic mutation density. Examining prostate biology through an epigenomic lens is fundamental for understanding the mechanisms underlying tumor progression.
Conflict of interest statement
Declaration of Interests
The authors declare no competing interests.
Figures
References
-
- Kunz M et al. RNA-seq analysis identifies different transcriptomic types and developmental trajectories of primary melanomas. Oncogene 37, 6136–6151 (2018). - PubMed
Methods-only References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
